Vrodené vývojové chyby plodu zaujímajú 2. – 3. miesto v štruktúre príčin perinatálnej smrti plodu a novorodenca. Veľký význam skorá diagnóza malformácií, ktorý je potrebný na včasné vyriešenie otázky možnosti predĺženia tehotenstva, ktorá je daná typom defektu, zlučiteľnosťou so životom a prognózou postnatálneho vývoja. V závislosti od etiológie sa rozlišujú dedičné (genetické), exogénne a multifaktoriálne vrodené malformácie plodu. Medzi dedičné patria malformácie vznikajúce mutáciami, t.j. pretrvávajúce zmeny v dedičných štruktúrach v gamétach alebo zygote. V závislosti od úrovne, na ktorej sa mutácia vyskytla (gény alebo chromozómy), sa rozlišujú monogénne syndrómy a chromozomálne ochorenia. Exogénne zahŕňajú defekty spôsobené škodlivým účinkom exogénnych faktorov. Tieto faktory, pôsobiace v období gametogenézy alebo tehotenstva, vedú k vzniku vrodených vývojových chýb bez narušenia štruktúry dedičného aparátu.
Malformácie multifaktoriálneho pôvodu sa nazývajú defekty, ktoré vznikli kombinovaným vplyvom genetických a exogénnych faktorov. Existujú aj izolované (lokalizované v jednom orgáne), systémové (v rámci jedného orgánového systému) a mnohopočetné (v orgánoch dvoch alebo viacerých systémov) defekty.
PORUCHY CENTRÁLNEHO NERVOVÉHO SYSTÉMU
Klasifikácia najčastejších malformácií centrálneho nervového systému:
1. Hydrocefalus:
Stenóza cerebrálneho akvaduktu;
otvorený hydrocefalus;
Dandy-Walkerov syndróm.
2. Papilóm choroidálneho plexu.
3. Poruchy neurálnej trubice:
- spina bifida;
anencefália;
Cefalokéla.
4. Mikrocefália. Hydrocefalus
Hydrocefalus- zväčšenie veľkosti mozgových komôr so súčasným zvýšením intrakraniálneho tlaku, sprevádzané vo väčšine prípadov zväčšením veľkosti hlavy (obr. 28).
Ryža. 28. Sonografický obraz ťažkého hydrocefalu plodu (šípky označujú ostro rozšírené mozgové komory, ktorých kôra je výrazne stenčená, veľkosť hlavičky plodu presahuje normálne hodnoty pre daný gestačný vek)
Ventrikulomegália je izolované zvýšenie veľkosti komôr, ktoré nie je sprevádzané zvýšením veľkosti hlavy. Hydrocefalus sa pozoruje s frekvenciou 0,1-2,5 na 1000 novorodencov. Asi 60 % plodov s hydrocefalom sú chlapci. Hydrocefalus môže byť výsledkom mnohých ochorení rôznej etiológie. Vo väčšine prípadov sa vyvíja v dôsledku porušenia odtoku cerebrospinálnej tekutiny. Komunikačná forma hydrocefalu je spôsobená extraventrikulárnym
kulárnej obštrukcie, zatiaľ čo obštrukčná forma je intraventrikulárna obštrukcia. Príležitostne vedie hydrocefalus k zvýšenej produkcii mozgovomiechového moku (napríklad na pozadí papilómu vaskulárnych plexusov) alebo k porušeniu jeho reabsorpcie v subarachnoidálnom priestore.
Extrakraniálne anomálie hydrocefalu sa vyskytujú v 63 %: agenéza a dysplázia obličiek, defekt komorového septa, Fallotova tetralógia, meningomyelokéla, štiepenie horná pera, mäkké a tvrdé podnebie, atrézia konečníka a konečníka, dysgenéza gonád. Hydrocefalus je reprezentovaný najmä stenózou mozgového akvaduktu (zúženie Sylviovho akvaduktu); otvorený hydrocefalus (rozšírenie komôr mozgu a subarachnoidálneho systému mozgu v dôsledku obštrukcie extraventrikulárneho systému odtokových ciest mozgovomiechového moku); Dandy-Walkerov syndróm (kombinácia hydrocefalu, cysty zadnej lebečnej jamy, defekty v cerebelárnom vermis, cez ktoré cysta komunikuje s dutinou IV komory). Keď sa zistí hydrocefalus, je potrebné starostlivo posúdiť anatómiu mozgových štruktúr, ako aj chrbtice, aby sa vylúčila spina bifida. Súčasťou komplexného vyšetrenia plodu by malo byť aj echokardiografické vyšetrenie, keďže hydrocefalus sa často kombinuje s vrodenými srdcovými chybami. Pri hydrocefale pred obdobím životaschopnosti plodu je vhodné prediskutovať otázku ukončenia tehotenstva s rodičmi. Pri predlžovaní tehotenstva sa dynamické ultrazvukové monitorovanie zobrazuje každé 2 týždne. S nárastom hydrocefalu po dosiahnutí zrelosti pľúc plodu by sa mala nastoliť otázka skorého pôrodu a posunu. Účinnosť prenatálneho ventrikulárneho bypassu zatiaľ nebola dokázaná a táto operácia nie je široko používaná. Cisársky rez je indikovaný len pri ťažkej makrocefálii a absencii iných malformácií. V prítomnosti hrubých kombinovaných anomálií, ktoré zhoršujú prognózu života, je operáciou voľby cefalocentéza.
Poruchy neurálnej trubice. Tento pojem zahŕňa anencefáliu, cefalokélu a spina bifida.
spina bifida- anomália vo vývoji chrbtice, ktorá je výsledkom porušenia procesu uzatvárania nervovej trubice (obr. 29).
Výstup cez defekt v membránach miechy sa nazýva meningokéla. Ak herniálny vak obsahuje nervové tkanivo, tvorba
 Ryža. 29. Sonografický obraz spina bifida v lumbosakrálnej chrbtici (označené šípkou)
Ryža. 29. Sonografický obraz spina bifida v lumbosakrálnej chrbtici (označené šípkou)
sa nazýva meningomyelokéla. Rozlišovať spina bifida cystica(cystická forma hernie chrbtice s tvorbou herniálneho vaku obsahujúceho membrány mozgu a/alebo substanciu mozgu) a spina bifida occulta(skrytá forma, ktorá nie je sprevádzaná tvorbou herniálneho výčnelku). Najčastejšie je táto chyba lokalizovaná v bedrovej a sakrálnej chrbtici. Frekvencia výskytu spina bifida závisí od geografického regiónu. V niektorých oblastiach Spojeného kráľovstva je frekvencia tohto defektu 4 na 1 000 novorodencov. V Spojených štátoch je toto číslo 0,5 na 1 000, hoci to závisí od rasových a geografických charakteristík. spina bifida- malformácia, ku ktorej dochádza v dôsledku porušenia uzáveru nervovej trubice v 4. týždni embryonálneho vývoja. Táto anomália sa dedí multifaktoriálne. rázštep chrbtice a môže vzniknúť v dôsledku hypertermie matky, ak má diabetes mellitus, vystavenie teratogénnym faktorom a môže byť súčasťou genetických syndrómov (s izolovaným mutantným génom) alebo chromozomálnych abnormalít (trizómia 13 a 18 párov chromozómov, triploidia , nevyvážená translokácia alebo kruhový chromozóm). Kýla chrbtice je spojená s viacerými
viac ako 40 syndrómov mnohopočetných malformácií (hydrocefalus, vrodené srdcové chyby a genitourinárny systém).
Prenatálne vyšetrenie zahŕňa stanovenie karyotypu a dôkladné ultrazvukové vyšetrenie. Osobitná pozornosť by sa mala venovať anatómii hlavy, srdca, rúk a nôh. Ak sa meningomyelokéla zistí pred životaschopnosťou plodu, žene by sa malo ponúknuť lekárske ukončenie tehotenstva. Pri predlžovaní tehotenstva je indikovaný dynamický ultrazvuk každé 2-3 týždne na posúdenie výskytu iných znakov (napríklad ventrikulomegália). Rodičom by mala byť poskytnutá konzultácia s neurochirurgom, aby prediskutovali možnosti chirurgického zákroku po pôrode (uzavretie defektu alebo bypass), ako aj prognózu života a zdravia dieťaťa. Pôrod by sa mal uskutočniť vo veľkých perinatálnych centrách, akonáhle pľúca plodu dosiahnu dostatočnú zrelosť. Empirické riziko recidívy spina bibida je 3-5%. Užívanie veľkých dávok kyseliny listovej (4 mg), ktoré sa začalo 3 mesiace pred plánovaným tehotenstvom a pokračovalo počas jeho prvej polovice, môže výrazne znížiť riziko defektu.
Akýkoľvek otvorený defekt neurálnej trubice by sa mal uzavrieť počas prvých 24 hodín života. Antibakteriálna terapia začatá hneď po narodení môže znížiť riziko infekčných komplikácií. Prognóza života a zdravia závisí od úrovne lokalizácie meningomyelokély, ako aj od počtu a povahy súvisiacich anomálií. duševný vývoj deti, ktoré majú pri narodení normálny obvod hlavy a správne vytvorený mozog, netrpia. Pacienti s meningomyelokélou lokalizovanou v L2 a vyššie sú takmer vždy nútení používať invalidný vozík.
Anencefália(pseudocefália, extrakraniálna dysencefália) - absencia mozgových hemisfér a väčšiny lebečnej klenby, pričom je defekt v čelovej kosti nad nadočnicovou oblasťou, chýba spánková a časť tylovej kosti. Horná časť hlavy je pokrytá cievnou membránou. Štruktúry stredného mozgu a diencefala sú čiastočne alebo úplne zničené. Hypofýza a kosoštvorcová jamka sú väčšinou zachované. Medzi typické prejavy patria vypúlené oči, veľký jazyk a krátky krk. Táto patológia sa vyskytuje s frekvenciou 1 z 1 000. Častejšie to
nájdené u novorodencov. Acrania(exencefália) - absencia lebečnej klenby v prítomnosti fragmentu mozgového tkaniva. Je to zriedkavejšia patológia ako anencefália. Anencefália je výsledkom neuzavretia rostrálneho neuropóru do 28 dní od oplodnenia. Zaznamenáva sa multifaktoriálna a autozomálne recesívna dedičnosť, chromozomálne abnormality. Medzi rizikové faktory patrí cukrovka matky. Pri pokusoch na zvieratách bola stanovená teratogenita žiarenia, salicylátov, sulfónamidov a zvýšených hladín oxidu uhličitého. Sonografickú diagnózu možno stanoviť už v 12-13 týždni tehotenstva. Anencefália a akrania sú absolútne smrteľné malformácie, preto v oboch prípadoch treba žene ponúknuť ukončenie tehotenstva. Všetci novorodenci s anencefáliou a akraniou zomierajú do 2 týždňov po narodení. Empirické riziko recidívy anencefálie je 3 – 5 %. Užívanie veľkých dávok kyseliny listovej (4 mg), ktoré sa začalo 3 mesiace pred plánovaným tehotenstvom a pokračovalo počas jeho prvej polovice, môže výrazne znížiť riziko defektu.
Cefalokéla(encefalokéla, kraniálna alebo okcipitálna meningokéla, rozštiepenie lebky) - vydutie obsahu lebky cez kostný defekt. Termín "kraniálna meningokéla" sa vzťahuje na výčnelok len cez defekt meningeálnych membrán. Keď je mozgové tkanivo v herniálnom vaku, používa sa termín "encefalokéla". Cefalokéla je zriedkavá (1:2000 živonarodených detí) a je súčasťou mnohých genetických (Meckeleho syndrómy, stredné rozštiepenie tváre) a negenetických (konstrikcia plodovej vody) syndrómov. Cefalokéla vzniká v dôsledku neuzavretia defektu neurálnej trubice a vyskytuje sa v 4. týždni vývoja. Defekt na lebke, cez ktorý môžu prolapsovať membrány mozgu a mozgového tkaniva, vzniká v dôsledku neoddelenia povrchového ektodermu a pod ním ležiaceho neuroektodermu. Ak sa zistí cefalokéla, žene má byť ponúknuté lekárske ukončenie tehotenstva. Pri predlžovaní tehotenstva závisí taktika pôrodu od veľkosti a obsahu herniálneho vaku. O veľké veľkosti defekt, prolaps značného množstva mozgového tkaniva, ako aj v prítomnosti mikrocefalie a hydrocefalu, prognóza pre život a zdravie je mimoriadne nepriaznivá.
Dodávka operáciou cisársky rez nezobrazené v týchto pozorovaniach. Je možné odporučiť dekompresiu herniálneho vaku, aby sa vytvorili podmienky pre pôrod cez prirodzené pôrodné cesty. Cisársky rez možno odporučiť, ak ide o malý defekt a ak je herniový vak malý.
Mikrocefália (mikroencefália) je klinický syndróm charakterizovaný zmenšením obvodu hlavy a mentálnou retardáciou. Vyskytuje sa s frekvenciou 1 z 1360 novorodencov, s kombinovanými anomáliami 1,6:1000 živonarodených detí. Mikrocefália je polyetiologické ochorenie, pri vzniku ktorého zohrávajú významnú úlohu genetické (chromozomálne aberácie, monogénne defekty) a faktory prostredia. Prognóza závisí od prítomnosti súvisiacich anomálií. Trizómia na chromozómoch 13, 18, Meckelov syndróm sú smrteľné lézie. Prenatálne vyšetrenie by malo zahŕňať stanovenie karyotypu plodu a dôkladné ultrazvukové vyšetrenie. Pri absencii sprievodných anomálií závisí prognóza od veľkosti hlavy: čím je menšia, tým nižší je index intelektuálneho rozvoja. Mikrocefália je nevyliečiteľná choroba. Pôrodnícka taktika - ukončenie tehotenstva pred dosiahnutím životaschopnosti plodu.
ANOMÁLIE ŠTRUKTÚR TVÁRIE A KRKU
Rázštep tváre(rázštep pery a podnebia) je lineárny defekt siahajúci od okraja pery k nosovému otvoru.
Rázštep podnebia v kombinácii s rázštepom pery sa cez alveolárne výbežky a tvrdé podnebie môže rozšíriť do nosnej dutiny alebo dokonca na dno očnice. Obojstranný rázštep pery sa pozoruje u 20%, rázštep pery a podnebia - 25%. Pri jednostrannej lézii je rázštep častejšie lokalizovaný vľavo. Rázštep tváre tvorí asi 13 % všetkých malformácií a zaznamenáva sa s frekvenciou 1:800 živonarodených detí. Chlapci majú rázštepy častejšie ako dievčatá. Kombinované anomálie sa nachádzajú v 50 % prípadov s izolovaným rázštepom podnebia a iba v 13 % s rázštepom pery a podnebia. Štruktúry tváre sa vytvárajú medzi 4. a 10. týždňom tehotenstva. Nepárové frontonazálne štruktúry sa spájajú s párovými maxilárnymi a mandibulárnymi štruktúrami.
moje tuberkulózy. Pri tých pozorovaniach, keď proces fúzie nie je dokončený, sa vytvárajú štrbiny. Spravidla je možné diagnostikovať rázštep tváre až v druhom trimestri tehotenstva skríningovým ultrazvukom. Prenatálna detekcia defektu pomocou echografie je náročná, no vďaka ultrazvukovému snímaniu a farebnému dopplerovskému mapovaniu sa rozširujú možnosti jej diagnostiky. Dopplerovský ultrazvuk dokáže vizualizovať pohyb tekutiny cez nos, ústa a hltan. V prítomnosti rázštepu sa mení charakter pohybu tekutiny. Trojrozmerná echografia môže objasniť diagnózu v tých pozorovaniach, keď bolo podozrenie na rázštep v dvojrozmernej štúdii, ale nezískala sa jeho jasná vizualizácia. Pomocou fetoskopie, vrátane embryonoskopie, je možné diagnostikovať anomáliu. Pri absencii pridružených anomálií sa používa všeobecne akceptovaná pôrodnícka taktika bez ohľadu na čas diagnózy. Užívanie kyseliny listovej pred a počas ďalšieho tehotenstva môže znížiť riziko rázštepov.
Rozštiepenie hornej pery (rázštep pery) nezasahuje do satia a je len kozmetickou vadou. Pri kombinácii rozštiepenia hornej pery, čeľuste a tvrdého podnebia (rázštep podnebia) sa zaznamenávajú funkčné poruchy: pri satí mlieko vyteká cez nos v dôsledku jeho komunikácie s ústnou dutinou; mlieko je možné inhalovať. Prognóza je priaznivá: moderné chirurgické metódy umožňujú dosiahnuť korekciu kozmetických a funkčných defektov.
cystický hygroma(lymfangióm alebo následky obštrukcie jugulárneho lymfatického kmeňa) je encystované nahromadenie tekutiny (obr. 30). Je charakterizovaná prítomnosťou jednej alebo viacerých cýst mäkkých tkanív na krku, ktoré sú výsledkom porúch lymfatického systému. Cystické hygromy sa vyskytujú s frekvenciou 1:200 spontánnych potratov (koccyx-parietálna veľkosť plodu nad 30 mm). Cystický hygrom sa často kombinuje s chromozomálnymi aberáciami (Turnerov syndróm, trizómie 13, 18, 21 párov chromozómov, mozaika). Ako izolovaná anomália sa dedí autozomálne recesívnym spôsobom. Prognóza: vo väčšine prípadov plod odumrie v prvých dvoch trimestroch tehotenstva. Asi 90 % vyžaduje chirurgickú liečbu, u 31 % sa objavia problémy s prehĺtaním a obštrukciou dýchacích ciest.
 Ryža. tridsať. Sonografický obraz cystického hygromu krčka plodu počas 16-týždňového tehotenstva (v krčku plodu je viditeľný veľký tekutý útvar - označený šípkou)
Ryža. tridsať. Sonografický obraz cystického hygromu krčka plodu počas 16-týždňového tehotenstva (v krčku plodu je viditeľný veľký tekutý útvar - označený šípkou)
spôsoby. Paréza tvárového nervu v dôsledku chirurgickej liečby sa vyskytuje u 24% pacientov.
Pôrodnícka taktika spočíva v ukončení tehotenstva s včasnou diagnózou cystického hygromu krku plodu, pri donosenom tehotenstve sa pôrod uskutočňuje prirodzeným pôrodným kanálom.
VRODENÉ SRDCE VADY
Frekvencia vrodených srdcových chýb (CHD) sa pohybuje od 1-2 do 8-9 na 1000 živonarodených detí. Najčastejšími ICHS sú defekty predsieňového a komorového septa, otvorený ductus arteriosus, stenóza pľúcnej tepny, syndróm hypoplastického ľavého srdca, jedna komora atď. V 90 % prípadov sú ICHS výsledkom multifaktoriálneho poškodenia (genetická predispozícia a faktory prostredia). Riziko recidívy defektu je 2-5% po narodení jedného a 10-15% po narodení dvoch chorých detí. Monogénna dedičnosť
starnutie sa pozoruje u 1-2% detí s vrodenou srdcovou chorobou. Chromozomálne abnormality sa nachádzajú u 5% detí, z ktorých trizómie sú hlavné. U 1-2% novorodencov sa vyskytuje kombinovaný účinok rôznych teratogénov. Fetálna echokardiografia je najinformatívnejšou metódou prenatálnej diagnostiky vrodených srdcových chorôb. Indikácie pre prenatálnu diagnostiku sú určené stavom matky a plodu.
1. Indikácie vzhľadom na stav matky:
Prítomnosť CHD u rodinných príslušníkov;
cukrovka;
Užívanie tehotných liekov počas organogenézy;
alkoholizmus;
systémový lupus erythematosus;
fenylketonúria.
2. Indikácie vzhľadom na stav plodu:
polyhydramnión;
neimunitná vodnateľnosť;
Poruchy srdcového rytmu;
Extrakardiálne defekty;
Chromozomálne poruchy;
Symetrická forma intrauterinnej retardácie rastu plodu. Prognóza závisí od typu defektu, prítomnosti sprievodných anomálií a chromozomálnych abnormalít.
Pôrodnícka taktika spočíva v tom, že po dôkladnej echokardiografickej štúdii sa vykoná cordo alebo amniocentéza, aby sa získal materiál na chromozomálnu analýzu. Ak sa ICHS zistí u neživotaschopného plodu, je indikované ukončenie tehotenstva. Pri donosenom tehotenstve je lepšie realizovať pôrod v špecializovaných perinatologických centrách. Pri kombinovaných defektoch a genetických abnormalitách je ukončenie tehotenstva nevyhnutné kedykoľvek.
jednej srdcovej komory. Ide o ťažkú vrodenú malformáciu, pri ktorej sú srdcové komory reprezentované jednou komorou alebo veľkou dominantnou komorou v kombinácii so spoločným atrioventrikulárnym spojením obsahujúcim dve atrioventrikulárne chlopne. Frekvencia výskytu defektu nebola presne stanovená. Jedna komora sa ľahko diagnostikuje pomocou štandardnej štvorkomorovej časti srdca plodu. Jediný
komora morfologicky môže byť pravá aj ľavá. Celková miera prežitia pre všetky typy jednej komory u pacientov bez chirurgickej liečby je 30 %. Jedna komora je často kombinovaná s chromozomálnymi abnormalitami, poruchami génov (Holt-Oramov syndróm), aspléniou/polyspléniou, často vznikajúcou pri niektorých ochoreniach matiek, a tiež na pozadí teratogénnych účinkov kyseliny retinovej. Prenatálne vyšetrenie pri náleze jednej komory by malo zahŕňať karyotypizáciu a podrobné vyšetrenie ultrazvukovej anatómie plodu. Klinický priebeh ochorenia a taktika manažmentu v novorodeneckom období sú určené stavom pľúcneho a systémového prietoku krvi.
Defekt predsieňového septa(AMPP) (obr. 31). Predstavuje nedostatok septa oddeľujúceho predsiene. Pozoruje sa u 17 % všetkých vrodených srdcových chýb a je jeho najčastejšou štrukturálnou anomáliou. Často v kombinácii s inými intrakardiálnymi anomáliami, ako aj neimunitnou vodnatosťou plodu. Možno kombinácia s chromozomálnymi abnormalitami. Väčšina malých ASD sa počas prenatálneho fetálneho ultrazvuku nezistí. Diagnózu možno vykonať iba pomocou viacerých rezov a farebného dopplerovského zobrazenia. Prenatálne vyšetrenie po zistení ASD by malo zahŕňať
 Ryža. 31. Sonografický obraz rozsiahleho defektu predsieňového septa (označený šípkou)
Ryža. 31. Sonografický obraz rozsiahleho defektu predsieňového septa (označený šípkou)
začať stanovenie karyotypu a podrobné štúdium ultrazvukovej anatómie plodu. Identifikácia izolovanej ASD v prenatálnom období nevyžaduje zmenu taktiky tehotenstva a pôrodu. IN neskoré termíny tehotenstva by sa malo vykonať dynamické hodnotenie stavu plodu.
Defekt komorového septa(VMZHP). Predstavuje nedostatok priehradky oddeľujúcej komory. Podľa lokalizácie sa rozlišujú defekty horného septa (na úrovni mitrálnej a trikuspidálnej chlopne), svalovej časti a výstupného úseku septa (subaortálny, subpulmonálny). Podľa veľkosti sú VSD rozdelené na malé (do 4 mm) a veľké. VSD môže byť izolovaná alebo kombinovaná s inými anomáliami, chromozomálnymi defektmi a dedičnými syndrómami. Vo všeobecnej štruktúre vrodených srdcových chýb je asi 20% izolovaných VSD, čo je najčastejšie diagnostikovaná chyba. Frekvencia malých, hemodynamicky nevýznamných, svalových defektov dosahuje 53:1000 živonarodených detí. Asi 90 % takýchto defektov sa spontánne uzavrie do veku 10 mesiacov a neovplyvňujú prognózu života a zdravia.
Väčšina malých VSD nie je detekovaná počas prenatálneho fetálneho ultrazvuku. Diagnózu možno vykonať iba pomocou viacerých rezov a farebného dopplerovského zobrazenia. Najčastejšie je VSD izolovaná, ale môže byť kombinovaná s chromozomálnymi abnormalitami, génovými poruchami, syndrómami viacerých malformácií. Prenatálne vyšetrenie na VSD by malo zahŕňať karyotypizáciu a podrobné vyšetrenie anatómie plodu ultrazvukom. Identifikácia izolovanej VSD v prenatálnom období nevyžaduje zmenu manažmentu tehotenstva a pôrodu. V neskorom tehotenstve by sa malo vykonať dynamické hodnotenie plodu. V prípade podozrenia na VSD je potrebné rodičom poskytnúť úplné informácie o prognóze života a zdravia nenarodeného dieťaťa a upovedomiť pediatra, aby zabezpečil adekvátne sledovanie novorodenca. Dokonca aj pri veľkých VSD môže byť ochorenie niekedy asymptomatické až 2-8 týždňov. V 50 % prípadov sa malé defekty spontánne uzavrú do 5. roku života a zo zvyšných 80 % vymiznú v r. dospievania. Väčšina pacientov s nekomplikovanou VSD má dobrú prognózu života a zdravia. S priaznivým prietokom
choroba významné obmedzenia fyzickej aktivity sa nevyžaduje.
Ebsteinova anomália- vrodená srdcová choroba, charakterizovaná abnormálnym vývojom a umiestnením cípov trikuspidálnej chlopne. Pri Ebsteinovej anomálii sa septálne a zadné plachty trikuspidálnej chlopne vyvíjajú priamo z endokardu pravej srdcovej komory, čo vedie k posunutiu abnormálnej chlopne hlboko do pravej komory a rozdeleniu komory na dve časti: distálnu (subvalvulárne) - aktívne a proximálne (supravalvulárne alebo atrializované) - pasívne. Supravalvulárne oddelenie, spojené s pravou predsieňou, tvorí jeden funkčný útvar. Ebsteinova anomália predstavuje 0,5 % všetkých vrodených srdcových chýb. Ebsteinovu anomáliu možno ľahko diagnostikovať vyšetrením štandardného štvorkomorového srdca plodu, pretože je takmer vždy sprevádzaná kardiomegáliou. Prenatálna diagnostika defektu je založená na detekcii výrazne zväčšeného pravého srdca v dôsledku pravej predsiene. Kľúčovým bodom v diagnostike Ebsteinovej anomálie je vizualizácia posunutej trikuspidálnej chlopne na pozadí rozšírenej pravej predsiene a normálneho myokardu pravej komory. Dôležitou prognostickou hodnotou pri Ebsteinovej anomálii je detekcia trikuspidálnej regurgitácie počas dopplerovskej echokardiografie plodu. Najskoršie prenatálne ultrazvuková diagnostika Ebsteinova anomália bola vykonaná v 18-19 týždni tehotenstva. Prognóza života s Ebsteinovou anomáliou je zvyčajne priaznivá v prípadoch, keď deti prežijú počas prvého roka života bez chirurgickej liečby. Ebsteinova anomália nie je často kombinovaná s chromozomálnymi aberáciami a syndrómami mnohopočetných vrodených chýb. Extrakardiálne anomálie sa pozorujú v 25%. Výsledok v novorodeneckom období závisí od závažnosti zmeny trikuspidálnej chlopne. Deti s ťažkou insuficienciou trikuspidálnej chlopne majú vysoké percento úmrtí. Klinicky sa insuficiencia trikuspidálnej chlopne prejavuje zvýšenou cyanózou, acidózou a príznakmi srdcového zlyhania. Chirurgická liečba je indikovaná u pacientov so závažnými príznakmi ochorenia, ktoré zasahujú do normálneho života dieťaťa. Operácia zahŕňa uzavretie septa
defekt, plasticita trikuspidálnej chlopne a jej pohyb na typické miesto. Úmrtnosť v nemocniciach je 6,3 %.
Fallotova tetralógia- komplexný defekt zahŕňajúci viaceré anomálie štruktúry srdca: defekt komorového septa, dextropozícia aorty, obštrukcia výstupnej pľúcnej tepny a hypertrofia pravej komory. Vo všeobecnej štruktúre vrodených srdcových chýb u živonarodených detí je Fallotova tetralógia 4-11%. Pri štúdiu štvorkomorového srdca plodu je veľmi ťažké diagnostikovať Fallotovu tetrádu. Pri prerezaní cez vývody hlavných tepien je možné vidieť typickú subaortálnu VSD a aortálnu dextrapozíciu. Dôležitým dodatočným kritériom je expanzia a posunutie koreňa aorty. Fallotova tetralógia je defekt modrého typu, t.j. u novorodencov sa výrazná cyanóza určuje vo veku 6 týždňov až 6 mesiacov. Fallotova tetralógia sa vzťahuje na ťažko diagnostikované srdcové chyby, ktoré často zostávajú neodhalené skríningovým ultrazvukom až do 22. týždňa tehotenstva. Najčastejšie je táto vada diagnostikovaná v treťom trimestri tehotenstva alebo po pôrode. Tetralogy of Fallot nevyžaduje špecifickú taktiku riadenia. Ak sa táto patológia zistí, je potrebné komplexné vyšetrenie a prenatálne poradenstvo. Takmer 30 % živonarodených detí s Fallotovou tetrádou má kombinované extrakardiálne anomálie. V súčasnosti je popísaných viac ako 30 syndrómov mnohopočetných malformácií, ktorých štruktúra zahŕňa Fallotovu tetralógiu. Prenatálne vyšetrenie na detekciu Fallotovej tetrády by malo zahŕňať stanovenie karyotypu a podrobnú štúdiu ultrazvukovej anatómie plodu. Prognóza života pri Fallotovej tetralógii do značnej miery závisí od stupňa obštrukcie výtokového traktu pravej komory. Viac ako 90 % pacientov, ktorí podstúpili kompletnú korekciu Fallotovej tetrády, sa dožije dospelosti. Z dlhodobého hľadiska sa 80 % pacientov cíti uspokojivo a má normálne funkčné parametre.
Transpozícia veľkých tepien- ochorenie srdca, pri ktorom aorta alebo jej väčšina vychádza z pravej komory a pľúcna tepna z ľavej komory. Tvorí 5-7% všetkých vrodených srdcových chýb. Zvyčajne nie je diagnostikovaná v prenatálnom období počas skríningu, pretože štúdium srdca plodu je obmedzené na štúdiu
iba štvorkomorový rez. Na identifikáciu defektu je potrebná vizualizácia hlavných ciev so štúdiom ich vzájomnej polohy. Normálne sa hlavné tepny krížia a počas transpozície opúšťajú komory paralelne: aorta - z pravej komory, pľúcna tepna - zľava. Transpozícia hlavných tepien s intaktnými medzikomorovými a interatriálnymi septami nie je zlučiteľná so životom. Asi 8 % živonarodených detí s transpozíciou hlavných tepien má pridružené extrakardiálne anomálie. Prenatálne vyšetrenie by malo zahŕňať stanovenie karyotypu a podrobnú štúdiu ultrazvukovej anatómie plodu. Väčšina novorodencov s transpozíciou hlavných tepien a intaktnou medzikomorovou priehradkou má výraznú cyanózu od prvých dní života. Chirurgická korekcia by sa mala vykonať hneď, ako sa zistí nedostatočné premiešanie prietokov krvi. Úmrtnosť novorodencov s týmto typom chirurgickej liečby je nižšia ako 5-10%.
CHOROBY HRUDNÍKA
vrodená diafragmatická hernia- defekt vyplývajúci zo spomalenia procesu uzatvárania pleuroperitoneálneho kanála. Pri tomto defekte býva nedostatočný rozvoj posterolaterálnej časti ľavej polovice bránice. Nedostatok oddelenia medzi brušnou dutinou a hrudníkom vedie k pohybu žalúdka, sleziny, čriev a dokonca aj pečene do hrudnej dutiny, čo môže byť sprevádzané posunom mediastína a spôsobiť stlačenie pľúc. V dôsledku toho sa často vyvíja bilaterálna pľúcna hypoplázia rôznej závažnosti. Nedostatočný rozvoj pľúc vedie k abnormálnej tvorbe ich cievneho systému a sekundárnej pľúcnej hypertenzii. Vrodená diafragmatická hernia sa vyskytuje približne u 1 z 2400 novorodencov.
Existujú štyri hlavné typy defektov: posterolaterálna (Bochdalekova hernia), anterolaterálna, sternálna a Morgagniho hernia. Obojstranné diafragmatické hernie tvoria 1 % všetkých typov defektov. Pohyb srdca na pravú stranu hrudník v kombinácii s echo-negatívnou štruktúrou (žalúdkom) v jeho ľavej polovici sa najčastejšie diagnostikuje ľavostranná diafragmatická hernia.
Pri pravostranných herniách je srdce zvyčajne posunuté doľava. V hrudníku sa dajú zobraziť aj črevá a pečeň. S touto chybou sa často zaznamenáva polyhydramnión. Kombinované anomálie sa pozorujú u 23 % plodov. Medzi nimi prevládajú vrodené srdcové chyby, ktoré tvoria 16 %. Diagnózu defektu možno vykonať už v 14. týždni tehotenstva. Úmrtnosť pri vrodenej diafragmatickej hernii koreluje s časom odhalenia defektu: iba 33 % novorodencov s defektom prežije v prípadoch, keď bola diagnóza stanovená pred 25. týždňom, a 67 % v prípade, že bola hernia zistená neskôr. Diafragmatické defekty majú väčšinou multifaktoriálny pôvod, avšak 12 % prípadov je kombinovaných s inými malformáciami alebo sú súčasťou chromozomálnych a nechromozomálnych syndrómov. Prenatálne vyšetrenie by malo nevyhnutne zahŕňať stanovenie karyotypu plodu a podrobné ultrazvukové vyšetrenie. Ak sa zistia kombinované anomálie, diferenciálnu diagnostiku možno vykonať iba počas konzultácie s genetikmi, syndromológmi a pediatrami. Rodičom by sa malo odporučiť, aby sa poradili s detským chirurgom, aby prediskutovali vlastnosti taktiky liečby v novorodeneckom období, prognózu života a zdravia. Priebeh novorodeneckého obdobia závisí od závažnosti pľúcnej hypoplázie a závažnosti hypertenzie. Veľký vplyv na výsledok v novorodeneckom období má aj veľkosť hernie a objem fungujúceho pľúcneho tkaniva. Abnormálny vývoj pľúc možno predpovedať pri polyhydramnióne, dilatácii žalúdka, ako aj pri pohybe pečene plodu do hrudnej dutiny. Podľa literatúry prežilo len 22 % detí diagnostikovaných prenatálne. Aj pri izolovanej vrodenej diafragmatickej hernii prežije len 40 %. Neonatálna smrť je zvyčajne výsledkom pľúcnej hypertenzie a/alebo respiračného zlyhania.
ANOMÁLIE TVORBY STENY brušnej dutiny A VÝVOJ VÝVOJA GASTROINTESTINAL TRACT
Omfalokéla (pupočná kýla)(obr. 32). Vyskytuje sa v dôsledku nevrátenia orgánov brušná dutina z amniovej dutiny cez pupočný krúžok. Omfalokéla môže zahŕňať akúkoľvek
 Ryža. 32. Sonografický obraz omfalokély (zobrazuje sa herniálny vak obsahujúci črevné kľučky a pečeň)
Ryža. 32. Sonografický obraz omfalokély (zobrazuje sa herniálny vak obsahujúci črevné kľučky a pečeň)
viscerálnych orgánov. Veľkosť herniálnej formácie je určená jej obsahom.
Je pokrytá amnioperitoneálnou membránou, pozdĺž ktorej bočnej plochy prechádzajú cievy pupočnej šnúry. Frekvencia omfalokély je 1 z 3000-6000 novorodencov. Existujú izolované a kombinované formy omfalokély. Táto patológia v 35-58% je sprevádzaná trizómiou, 47% - vrodenými srdcovými chybami, 40% - malformáciami genitourinárneho systému, 39% - defektmi neurálnej trubice. V 20% prípadov sa zistí intrauterinná rastová retardácia.
Prenatálna ultrazvuková diagnostika je založená na detekcii okrúhleho alebo oválneho útvaru vyplneného brušnými orgánmi a priliehajúceho priamo k prednej brušnej stene. Najčastejšie zloženie herniálneho obsahu zahŕňa črevné slučky a pečeň. Pupočná šnúra je pripevnená priamo k herniálnemu vaku. V niektorých prípadoch môže byť prenatálna diagnóza stanovená na konci prvého trimestra tehotenstva, hoci vo väčšine prípadov sa omfalokéla zistí v druhom trimestri. Prognóza závisí od sprievodných anomálií. Perinatálne straty sú častejšie spojené s ICHS, chromozomálne
aberácie a predčasnosť. Najväčší defekt sa odstraňuje jednostupňovou operáciou, pri veľkej sa robia viacstupňové operácie na uzavretie otvoru v prednej brušnej stene silikónovou alebo teflónovou membránou. Pôrodnícka taktika je určená obdobím detekcie defektu, prítomnosťou kombinovaných anomálií a chromozomálnych porúch. Ak sa defekt zistí v počiatočných štádiách tehotenstva, je potrebné ho prerušiť. V prípade zistenia sprievodných anomálií nezlučiteľných so životom je potrebné tehotenstvo kedykoľvek ukončiť. Spôsob pôrodu závisí od životaschopnosti plodu, keďže pri pôrode s veľkou omfalokélou môže dôjsť k prasknutiu herniálneho vaku a infekcii vnútorných orgánov plodu.
Gastroschíza- defekt prednej brušnej steny v oblasti pupka s eventeráciou črevných kľučiek pokrytých zápalovým exsudátom. Defekt sa zvyčajne nachádza vpravo od pupka, herniálne orgány nemajú membránu. Frekvencia gastroschízy je 0,94:10 000 novorodencov. Frekvencia defektu u tehotných žien mladších ako 20 rokov je vyššia a je 7 na 10 000 novorodencov.
Od konca 70. rokov. 20. storočie v Európe a Spojených štátoch pokračuje trend zvyšovania frekvencie pôrodov detí s gastroschízou. Prideľte izolované a kombinované formy. Izolovaná gastroschíza je bežnejšia a predstavuje v priemere 79 %. Kombinovaná forma sa zistí v 10-30% prípadov a najčastejšie ide o kombináciu gastroschízy s atréziou alebo črevnou stenózou. Medzi ďalšie anomálie, vrodené chyby srdca a močového systému, syndróm sušené slivky, hydrocefalus, nízky a polyhydramnión.
Anomália sa vyskytuje sporadicky, existujú však pozorovania rodinného ochorenia s autozomálne dominantným typom dedičnosti.
Najskoršia prenatálna ultrazvuková diagnostika pomocou transvaginálnej echografie sa uskutočnila v 12. týždni tehotenstva. Vo väčšine prípadov je diagnóza stanovená v druhom trimestri tehotenstva, pretože v počiatočných štádiách (10-13 týždňov) je možná falošne pozitívna diagnóza v dôsledku prítomnosti fyziologickej črevnej hernie u plodu. Prenatálna ultrazvuková diagnostika gastroschízy je zvyčajne založená na vizualizácii črevných slučiek v plodová voda v blízkosti prednej brušnej steny plodu. Niekedy, okrem črevných slučiek, mimo
iné orgány môžu byť umiestnené v brušnej dutine. Presnosť ultrazvukovej diagnostiky gastroschízy v II a III trimestri gravidity sa pohybuje od 70 do 95 % a závisí od gestačného veku, polohy plodu, veľkosti defektu a počtu orgánov umiestnených mimo prednej brušnej steny. .
Celková prognóza pre novorodencov s izolovanou gastroschízou je dobrá, pričom viac ako 90 % dojčiat prežije. Pri predlžovaní tehotenstva nemá taktika vedenia v druhom trimestri žiadne špeciálne vlastnosti. Vzhľadom na nízku frekvenciu kombinácie izolovanej gastroschízy s chromozomálnymi abnormalitami je možné upustiť od prenatálnej karyotypizácie. V treťom trimestri tehotenstva je potrebné vykonať dynamické hodnotenie funkčného stavu plodu, pretože frekvencia úzkosti pri gastroschíze je pomerne vysoká a retardácia intrauterinného rastu sa tvorí v 23 - 50% prípadov.
Ak sa gastroschíza zistí pred životaschopnosťou plodu, je potrebné vykonať potrat. Pri donosených tehotenstvách sa pôrod vykonáva v inštitúcii, kde je možné poskytnúť chirurgickú starostlivosť.
duodenálna atrézia je najčastejšou príčinou obštrukcie tenkého čreva. Frekvencia anomálie je 1:10 000 živonarodených detí. Etiológia nie je známa. Je možný výskyt defektu pod vplyvom teratogénnych faktorov. Sú opísané rodinné pozorovania pyloroduodenálnej atrézie s autozomálne recesívnou dedičnosťou. U 30 – 52 % pacientov je anomália izolovaná a u 37 % sú zistené malformácie kostrového systému: abnormálny počet rebier, agenéza krížovej kosti, konská noha, obojstranné krčné rebrá, obojstranná absencia prvých prstov , atď. U 2% sú diagnostikované kombinované anomálie gastrointestinálneho traktu: neúplná rotácia žalúdka, atrézia pažeráka, ilea a konečníka, transpozícia pečene. U 8-20% pacientov sa zistia vrodené srdcové chyby, približne v 1/3 prípadov sa atrézia dvanástnika kombinuje s trizómiou pre 21 párov chromozómov. Hlavnými prenatálnymi echografickými nálezmi v duodenálnej atrézii sú polyhydramnión a klasický znak "dvojitá bublina" v brušku plodu. Obraz "dvojitej bubliny" sa objavuje v dôsledku rozšírenia časti dvanástnika a žalúdka. Zovretie medzi týmito formáciami je tvorené pylorickou časťou žalúdka
ka a má veľký význam pre presnú prenatálnu diagnostiku tejto vady. V prevažnej väčšine prípadov je duodenálna atrézia diagnostikovaná v II a III trimestri tehotenstva. V skorších termínoch predstavuje diagnostika tohto defektu značné ťažkosti. Najskoršia diagnóza duodenálnej atrézie bola stanovená v 14. týždni.
Na určenie pôrodníckej taktiky sa vykonáva podrobné ultrazvukové hodnotenie anatómie vnútorných orgánov plodu a jeho karyotypizácia. Pred začiatkom obdobia životaschopnosti plodu je indikované ukončenie tehotenstva. Ak sa v treťom trimestri zistí izolovaná anomália, je možné predĺženie gravidity, po ktorom nasleduje pôrod v regionálnom perinatálnom centre a chirurgická korekcia malformácie.
Izolovaný ascites. Ascites je nahromadenie tekutiny v peritoneálnej dutine. Frekvencia nebola presne stanovená. Pri ultrazvukovom vyšetrení plodu sa ascites prejavuje prítomnosťou echo-negatívneho priestoru s hrúbkou 5 mm a viac v brušnej dutine. V prenatálnom období môže byť ascites izolovaný alebo môže byť jedným zo znakov vodnatosti neimunitného pôvodu. Okrem ascitu je vodnateľnosť plodu charakterizovaná prítomnosťou subkutánneho edému, pleurálnych a perikardiálnych výpotkov, ako aj nárastom hrúbky placenty o viac ako 6 cm, polyhydramniónom a hydrokélou.
Ascites sa môže kombinovať s rôznymi štrukturálnymi anomáliami, preto je indikované dôkladné vyšetrenie všetkých vnútorných orgánov plodu. Medzi príčinami izolovaného ascitu je potrebné rozlišovať mekóniovú peritonitídu a vrodenú hepatitídu.
Doteraz v literatúre neexistujú žiadne publikácie o detekcii izolovaného ascitu v prvom trimestri gravidity. Väčšina pozorovaní včasnej diagnostiky ascitu sa vyskytuje na začiatku druhého trimestra tehotenstva. Jednou z najčastejších príčin neimunitnej vodnatieľky sú chromozomálne abnormality. Pri izolovanom ascite sú chromozomálne defekty menej časté, ale musia sa brať do úvahy ako možné pozadie rozvoja tejto patológie. Pri zistení ascitu u plodu je najprv potrebné vylúčiť kombinované anomálie a intrauterinné infekcie. Priebeh ascitu plodu závisí od jeho etiológie. Idiopatický izolovaný ascites má priaznivú prognózu. Vo viac ako 50 % pozorovaní je zaznamenané jeho spontánne vymiznutie. Najčastejšou príčinou izolovaného ascitu je intrauterinná infekcia.
parvovírus B19. Pri predlžovaní tehotenstva je potrebné vykonať dynamické echografické pozorovanie vrátane dopplerovského hodnotenia prietoku krvi v žilovom kanáliku. Pri normálnych hodnotách prietoku krvi v žilovom kanáliku u plodov s ascitom je vo väčšine prípadov zaznamenaný priaznivý perinatálny výsledok. S nárastom ascitu niektorí autori odporúčajú terapeutickú punkciu, najmä v prípadoch, keď proces prebieha v neskorom tehotenstve. Hlavným účelom punkcie je zabrániť diskoordinácii pracovná činnosť a respiračné ťažkosti v novorodeneckom období. Ak sa v prenatálnom období zistí izolovaný ascites a vylúči sa komorbidita nezlučiteľná so životom, dieťa potrebuje po pôrode starostlivé dynamické sledovanie a symptomatickú terapiu.
Malformácie obličiek a močových ciest
Renálna agenéza- úplná absencia oboch obličiek. Výskyt defektu je spôsobený porušením sekvenčného reťazca procesov normálnej embryogenézy od pronephros po metanefros. Frekvencia je v priemere 1:4500 novorodencov. Je potrebné poznamenať, že u chlapcov sa vyskytuje dvakrát častejšie. Patognomická triáda echografických príznakov agenézy obličiek u plodu je reprezentovaná absenciou ich ozveny a močového mechúra, ako aj ťažkým oligohydramniónom. Oligohydramnios sa vzťahuje na neskoré prejavy a možno ho zistiť po 16-18 týždni tehotenstva. Zvyčajne je bilaterálna agenéza obličiek sprevádzaná symetrickou formou syndrómu retardácie rastu plodu. Agenéza obličiek je najčastejšie sporadická, ale môže sa kombinovať s rôznymi anomáliami vnútorných orgánov. Priamymi dôsledkami oligohydramniónu sú pľúcna hypoplázia, deformácie skeletu a tváre, syndróm retardácie rastu plodu. Renálna agenéza bola opísaná pri viac ako 140 syndrómoch mnohopočetných vrodených malformácií, chromozomálnych abnormalít a teratogénnych účinkov. Po diagnostikovaní by sa mala karyotypizácia vykonať prenatálne alebo po narodení, aby sa vylúčili chromozomálne abnormality. Pri všetkých pozorovaniach agenézy obličiek je potrebné kompletné patoanatomické vyšetrenie. Je zobrazená ultrasonografia
vyšetrenie obličiek u najbližších príbuzných. Pri prenatálnom zistení defektu by sa malo kedykoľvek odporučiť prerušenie tehotenstva. Ak sa rodina rozhodne predĺžiť tehotenstvo, je indikovaná konzervatívna pôrodnícka taktika.
Autozomálne recesívne polycystické ochorenie obličiek (infantilná forma). Prejavuje sa obojstranným symetrickým zväčšením obličiek v dôsledku náhrady parenchýmu sekundárne zväčšeným zberným kanálikom bez proliferácie väziva. Pohybuje sa od klasického smrteľného variantu až po infantilné, juvenilné a dokonca aj dospelé formy. V infantilnej forme dochádza k sekundárnej dilatácii a hyperplázii normálne vytvorených zberných kanálikov obličiek. Obličky sú ovplyvnené symetricky, zatiaľ čo cystické útvary majú veľkosť 1-2 mm. Frekvencia je 1,3-5,9:1000 novorodencov. Hlavnými echografickými kritériami pre malformáciu sú zväčšené hyperechoické obličky, absencia ozveny močového mechúra a oligohydramnión. Zväčšenie veľkosti obličiek je niekedy také výrazné, že zaberajú veľkú časť prierezu plodového brucha. Typický echografický obraz sa môže objaviť až v treťom trimestri tehotenstva. Prognóza je nepriaznivá. Smrť pochádza zo zlyhania obličiek. Pôrodnícka taktika je kedykoľvek prerušiť tehotenstvo.
Polycystické ochorenie obličiek u dospelých(autozomálne dominantné ochorenie, hepatorenálna polycystická choroba dospelého typu, Potterov syndróm III. typu) je charakterizovaná náhradou obličkového parenchýmu početnými cystami rôznych veľkostí, ktoré vznikajú v dôsledku rozšírenia zberných kanálikov a iných tubulárnych segmentov nefrón. Obličky sú ovplyvnené na oboch stranách a zväčšené, ale jednostranný proces môže byť prvým prejavom ochorenia. Na patologickom procese sa podieľa aj pečeň - vzniká periportálna fibróza, ktorá má ohniskový charakter. Etiológia ochorenia nie je známa, ale typ dedičnosti spôsobuje 50% riziko vzniku ochorenia a jeho genetické zameranie sa nachádza na 16. páre chromozómov. Mutantný gén je nositeľom jedného z 1000 ľudí. K penetrácii génov dochádza v 100 % prípadov, priebeh ochorenia sa však môže líšiť od ťažkých foriem s fatálnym koncom v novorodeneckom období až po asymptomatické formy zistené až pri pitve.
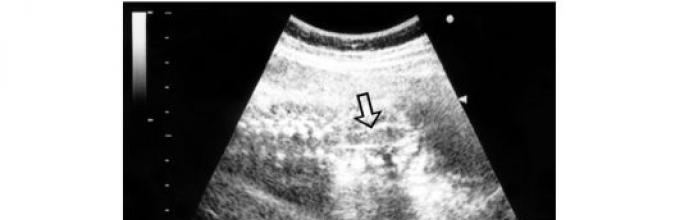
Polycystické ochorenie obličiek(multicystické ochorenie, cystické ochorenie obličiek, Potterov syndróm typu II, dysplastické ochorenie obličiek) je charakterizované cystickou degeneráciou renálneho parenchýmu v dôsledku primárnej expanzie renálnych tubulov. Pri multicystickej dysplázii obličiek sú ureter a panva najčastejšie atrezované alebo chýbajú. Proces môže byť bilaterálny, jednostranný a segmentový. Pri multicystickej dysplázii je oblička zvyčajne výrazne zväčšená; chýba obvyklý tvar a normálne tkanivo. Oblička je reprezentovaná mnohopočetnými cystami s anechogénnym obsahom (obr. 33).
 Ryža. 33. Echogram bilaterálnych polycystických obličiek plodu (ostro zväčšené obličky obsahujúce viaceré cysty rôznych priemerov - označené šípkou)
Ryža. 33. Echogram bilaterálnych polycystických obličiek plodu (ostro zväčšené obličky obsahujúce viaceré cysty rôznych priemerov - označené šípkou)
Veľkosti cýst sa líšia v pomerne širokom rozmedzí a závisia od trvania tehotenstva. Bližšie k termínu môže priemer cýst dosiahnuť 3,5-4 cm.Močový mechúr je zvyčajne vizualizovaný jednostranným procesom a nie je vizualizovaný bilaterálnym procesom. Pri bilaterálnom procese sa zvyčajne zaznamenáva oligohydramnión. Ochorenie sa vyskytuje prevažne sporadicky a môže byť sekundárne v kombinácii s inými syndrómami. pôrodníckeho
taktika v bilaterálnom procese diagnostikovaná v skorých štádiách, vzhľadom na nepriaznivú prognózu, je prerušenie tehotenstva. Pri jednostrannom procese a normálnom karyotype bez pridružených anomálií je indikovaný klasický pôrod s následnou konzultáciou dieťaťa s odborníkom.
Rozšírenie močových ciest. Môžu byť spôsobené anomálie genitourinárneho systému plodu sprevádzané rozšírením močových ciest rôzne dôvody, vrátane vezikoureterálneho refluxu, idiopatickej pyelektázy, obštrukčných porúch a pod. Z klinického hľadiska je vhodné odlíšiť pyelektázu a obštrukčnú uropatiu v prenatálnom období.
Pyelectáza. Pyelectáza je charakterizovaná nadmernou akumuláciou tekutiny a expanziou obličkovej panvičky plodu.
Pyelectáza je najčastejším nálezom na ultrazvuku plodu. Frekvencia jeho vývoja nebola stanovená, pretože táto patológia je sporadický jav. Chlapcom sa po narodení diagnostikuje 5-krát častejšie. U 27 % detí s hydronefrózou sa zistí vezikoureterálny reflux, obojstranné zdvojenie močovodov, obojstranný obštrukčný megaureter, nefunkčná kontralaterálna oblička a jej agenéza, u 19 % anomálie vo vývoji rôznych orgánov. Na prenatálnu ultrazvukovú diagnostiku pyelectázy by sa mali obličky plodu vyšetriť pri priečnom aj pozdĺžnom skenovaní. Pri priečnom snímaní obličky sa dilatácia obličkovej panvičky posudzuje na základe jej predo-zadnej veľkosti. Väčšina výskumníkov považuje pyelectázovú expanziu obličkovej panvičky v druhom trimestri tehotenstva o viac ako 5 mm a v trimestri III o viac ako 8 mm. Pri rozšírení obličkovej panvičky plodu nad 10 mm sa zvykne hovoriť o hydronefróze. Najbežnejšia klasifikácia hydronefrózy u plodu je:
Stupeň I (fyziologická dilatácia):
Renálna panva: predo-zadný rozmer<1 см;
Kortikálna vrstva: nezmenená.
Stupeň II:
Renálna panvička: 1,0-1,5 cm;
Poháre: nezobrazené;
Kortikálna vrstva: nezmenená.
Stupeň III:
Renálna panvička: predozadný rozmer >1,5 cm;
Kalichy: mierne rozšírené;
Kortikálna vrstva: nezmenená.
Stupeň IV:
Renálna panvička: predozadný rozmer >1,5 cm;
Kalich: stredne rozšírený;
Kortikálna vrstva: mierne zmenená.
Stupeň V:
Renálna panvička: predozadný rozmer >1,5 cm;
Kalichy: značne rozšírené;
Kortikálna vrstva: atrofia.
Rozšírenie obličkovej panvičky plodu možno pozorovať s rôznymi chromozomálnymi abnormalitami. Frekvencia chromozomálnych defektov u plodov s pyeloektáziou je v priemere 8 %. U väčšiny plodov s chromozomálnymi abnormalitami sa zistí kombinácia pyelectázy a iných vývojových anomálií. Stredne výrazná pyelectáza má dobrú prognózu a potreba chirurgickej liečby po pôrode sa vyskytuje pomerne zriedkavo. Vo väčšine pozorovaní je zaznamenané spontánne vymiznutie stredne ťažkej pyelectázy po narodení dieťaťa.
Pôrodnícka taktika závisí od času výskytu a trvania priebehu patologického procesu, ako aj od stupňa poškodenia funkcie obličiek. Predčasný pôrod je odôvodnený oligohydramniónom. V postnatálnom období je zobrazené dynamické sledovanie a konzultácia s detským urológom.
obštrukčná uropatia. Obštrukciu močových ciest u plodu možno pozorovať na akejkoľvek úrovni: vysoká obštrukcia, obštrukcia na úrovni ureteropelvickej fistuly (PUR), obštrukcia na strednej úrovni (ureter), obštrukcia na úrovni vezikoureterálneho spojenia (VUR) , nízka obštrukcia (uretra). URMS je najčastejšou príčinou obštrukčnej uropatie u plodu a tvorí v priemere 50 % všetkých vrodených urologických anomálií. Medzi hlavné sonografické znaky OLMS patrí dilatácia obličkovej panvičky s alebo bez expanzie kalichov; močovody nie sú vizualizované; močový mechúr môže mať normálnu veľkosť alebo v niektorých prípadoch nemusí byť vizualizovaný. Taktika v OLMS by mala byť očakávaná. Inštalácia veziko-amniotického skratu nie je indikovaná. Na ultrazvukový plač -
OPMS u plodu zahŕňa dilatáciu močovodu a pyelektázu. Močový mechúr má zvyčajne normálnu veľkosť. Taktika vedenia je podobná ako v OLMS. Najčastejšou príčinou nízkej obštrukcie sú chlopne zadnej uretry. Pri ťažkej obštrukcii sa pozoruje oligohydramnión, ktorý vedie k hypoplázii pľúc, deformáciám tvárových štruktúr a končatín, fibróze a dysplázii parenchýmu obličiek. Echografický obraz je charakterizovaný prítomnosťou dilatovanej uretry proximálne od miesta obštrukcie, výraznou expanziou močového mechúra. Prenatálny manažment nízkej obštrukcie závisí od dĺžky tehotenstva, prítomnosti oligohydramniónu a súvisiacich anomálií a od funkčného stavu obličiek. Pri stredne závažnej a neprogresívnej pyelectáze by sa mala dodržiavať konzervatívna taktika. Pri progresii obštrukčných porúch je opodstatnený pôrod s možnou chirurgickou korekciou defektu, aby sa predišlo závažným poruchám obličiek u plodu. Pri predčasnom tehotenstve u plodov s ťažkou obštrukčnou uropatiou možno vykonať intrauterinnú chirurgickú korekciu defektu.
VADY KOSTÍ
Spomedzi vrodených malformácií kostrového systému sa najčastejšie vyskytuje amélia (aplázia všetkých končatín); fokomélia (nedostatočný rozvoj proximálnych končatín, zatiaľ čo ruky a nohy sú spojené priamo s telom); aplázia jednej z kostí dolnej časti nohy alebo predlaktia; polydaktýlia (zvýšenie počtu prstov na končatine); syndaktýlia (zníženie počtu prstov v dôsledku fúzie mäkkých tkanív alebo kostného tkaniva susedných prstov); abnormálne nastavenie zastavenia; osteochondrodysplázia, charakterizovaná anomáliami v raste a vývoji chrupavky a / alebo kostí (achondrogenéza, achondroplázia, thanatoformná dysplázia, osteogenesis imperfecta, hypofosfatázia atď.).
Najdôležitejšia je diagnostika defektov nezlučiteľných so životom. Mnohé formy skeletálnej dysplázie sa kombinujú s hypopláziou pľúc v dôsledku malej veľkosti hrudníka v dôsledku nedostatočného rozvoja rebier. Vývoj pľúcnej insuficiencie v tomto prípade môže byť príčinou smrti detí v prvých hodinách mimomaternicového života.
Achondroplázia je jednou z najčastejších neletálnych kostných dysplázií a v 90 % prípadov je spôsobená novou mutáciou. Achondroplázia je osteochondroplázia s defektmi v dlhých kostiach a/alebo axiálnom skelete. Frekvencia je 0,24-5:10 000 pôrodov. Pomer mužských a ženských plodov je 1:1. Skrátenie kostí pri achondroplázii sa u plodu môže objaviť až v 24. týždni tehotenstva. Klasické echografické nálezy zahŕňajú krátke končatiny (menej ako 5. percentil), malý hrudník, makrocefáliu a sedlový nos. Priemerná dĺžka života s achondropláziou závisí predovšetkým od času, keď malá veľkosť hrudníka nespôsobuje vážne problémy s dýchaním. Intelektuálny vývoj defektu je normálny, existuje však vysoké riziko neurologických porúch, najmä kompresie miechy na úrovni foramen magnum, čo môže obmedzovať fyzické cvičenie. Makrocefalia môže byť výsledkom mierneho hydrocefalu v dôsledku malej veľkosti foramen magnum. Achondroplázia je dobre študovaný a bežný typ vrodeného trpaslíka u novorodencov. Vážne problémy môžu mať centrálne a obštrukčné spánkové apnoe. Vo veku 6-7 rokov života sa často zaznamenávajú chronické recidivujúce infekcie stredného ucha. V ranom detstvačasto sa vyskytuje aj zakrivenie dolných končatín, ktoré si pri ťažkých stavoch vyžaduje chirurgickú korekciu. Zvyčajne sa výška dospelých s achondropláziou pohybuje od 106 do 142 cm.
AHOJ! SOM V POZÍCII 32-33 TÝŽDŇA. PRI POSLEDNOM ULTRAZVUKOVOM OBJEME CNS PLODU: HYDROCEFÁLIA, AGENÉZIA CALLOSUM, ARCHOIDÁLNA CYST. MALÉ VSD V PLODE. POČAS TEHOTENSTVA SOM BOLA 2x V NEMOCNICI: TERMÍN 6-7 TÝŽDŇA A 28-29 TÝŽDŇA S HROZENÍM PREDČASNÉHO PÔRODU. POČAS TEHOTENSTVA BOLI USKUTOČNENÉ 3 skríningy, KDE SA PLODU VYVINULO NORMÁLNE BEZ INDIKÁTOROV CNS CM ATD. LEN V 31-32 TÝŽDNI SOM OBJISTIL TAKÚ DIAGNOSTIKU, KTORÚ POTVRDILI 3 NÁS ŠPECIALISTI. V UTERINOVEJ DUTINE 1 PLOD V PREZENTÁCII HLAVY. FETOMETRIA: BRG-79MM(32WEEKS) OG-285MM(31N.2DAY) OJ 257MM(31N.) DĹŽKA PRAVEJ A ĽAVEJ HUMERU 54MM-31WEEK. 6D. DĹŽKA PRAVEJ A ĽAVEJ SENNEJ 56MM.-29TÝŽDŇA 5 DNÍ DL. ATĎ. A LEV. HOLENNÉ KOSTI 51MM.-31TÝŽ DL. RÁDIUM KOST 44MM.- 30 TÝŽDŇOV 6 DNÍ ODHADOVANÁ HMOTNOSŤ OVOCIA 1520 -+ 210GR. ANATÓMIA PLODU: BOČNÉ KOMORY MOZGU SÚ ROZŠÍRENÉ, ASYMETRICKÉ, ROHY BOČNÝCH KOMOR VĽAVO 17MM, PRAVÉ 26MM. TRETIA KOMORA JE DILATANÁ NA 10 MM. DUTINA TRANSPARENTNEJ PRIESKY NIE JE VIZUALIZOVANÁ. V SAGITÁLNEJ SEKCI NIE JE CALLOSUM VÝZNAMNE UMIESTNENÉ. NA STRANE STREDNEJ ČIARY, VĽAVO, V RÁMCI PREDNÝCH A STREDNÝCH POZÍCIÍ LEBEKY SA NACHÁDZA ÚTVOR ANECHOGENICKEJ ŠTRUKTÚRY okrúhleho tvaru VEĽKOSTI 40*28 MM, AVASKULÁRNY V REŽIME CDI. VEĽKÁ NÁDRŽ 6 MM. INTERHEMISFÉROVÁ VEĽKOSŤ CERENERLA 36 MM - 30 TÝŽDŇOV.3 DNI. SYLVIAN'S FUROUS SA NACHÁDZA. ŠTRUKTÚRY TVÁRE: PROFIL - BEZ VLASTNOSTÍ, NOSOLABICKÝ TROJUHOLNÍK - BEZ VLASTNOSTÍ. ŠÍRKA ALVEOLÁRNEHO PROCESU 27 MM. PRIEMER OKA 15 MM, VNÚTRORBITÁLNY ROZMER 17,5 MM. CHRBTA BEZ DEFORMÁCIE. PĽÚCA BEZ VLASTNOSTÍ. Ozveny PLODU. VRCHNÁ ČASŤ SRDCA SA NACHÁDZA V ĽAVOM PREDNOM KVADRANTE Hrudnej dutiny, ZÁKLADNA RVF JE VĽAVO OD STERNUM. VEĽKOSTI SRDCA PRAVIDELNÉ. Srdcová frekvencia 138 úderov. V MIN. OS SRDCA V UHLE 45 STUPŇOV TÝKAJÚCE SA SAGITNEJ ROVINY. VEĽKOSTI ÁTRIA SÚ ROVNAKÉ, VENTIL OVÁLNEJ FORMY OTVORENÝ DO DUTINY ĽAVÉHO ÁTRIA, FUNKCIE. VEĽKOSTI KOMORY SÚ ROVNAKÉ. V STREDNEJ TRETINE MR JE ECHO-NEGATÍVNA ZÓNA 1MM. ATRIVENTRIKULÁRNE VENTILY SÚ UMIESTNENÉ NA ROVNAKEJ ÚROVNI A FUNGUJÚ SYNCHRONÓZNE. ŠTRUKTÚRA MYOKARDY, ENDOKARDIE A PERIKARDIE JE NORMÁLNA. KRÁTKE OSI ÚSEKY CEZ HLAVNÉ TEPINY A PREREZ CEZ 3 CIEVY BEZ VLASTNOSTÍ. V REŽIME CFM VO VZOSTUPNEJ AORTE A PĽÚCNE TEPINY LAMINÁT. ŽALÚDOK, ŽLČNÍK, ČREVÁ, OBLIČKY, PEČEŇ- BEZ VLASTNOSTÍ. MIESTO PRIPOJENIA PPUPOČNÍKA K PREDNEJ BRUŠNEJ STENE SA NACHÁDZA. PLACENTA SA NACHÁDZA NA PREDNEJ STENE MATICE, ZASAHUJE DNO. HRÚBKA PLACENTY NORMÁLNA-33MM. STUPEŇ ZRELOSTI JE 1., KTORÝ ZODPOVEDÁ DÁTUMU TEHOTENSTVA. MNOŽSTVO OKOLITEJ VODY JE NORMÁLNE. PPUPOČNÁ ŠNÚRA MÁ 3 CIEVY, UPOJENÉ PARACENTRÁLNE. STENY UTERUS BEZ VLASTNOSTÍ. PORUŠENIE UTERO-PLACENTÁLNEHO A PLACENTÁLNEHO PRÚDU KRVI PLODU SA NEZISTÍ. ČO MÁ TOTO DIEŤA, ČO MÁM OČAKÁVAŤ A ČO ROBIŤ?
Vrodené malformácie plodu zahŕňajú nasledujúce anomálie, ktoré sa vyskytujú u nenarodeného dieťaťa:
Neprítomnosť mozgu (anencefália);
- otvorená forma hernie miechy (back bifida);
- vrodené malformácie močového systému plodu;
- srdcové ochorenie u plodu alebo patologické zmeny v kardiovaskulárnom systéme;
- rôzne anomálie vo vývoji končatín u plodu - atrézia (absencia končatín);
- rázštep pery a podnebia, iné maxilofaciálne deformity.
Prečo sa vyskytujú vrodené malformácie plodu
Vznik a vývoj rôznych defektov u plodu môže nastať ako dôsledok vplyvu veľkého množstva faktorov, z ktorých väčšina zostala doteraz neobjasnená.
Podľa etiologických znakov sú všetky vrodené malformácie plodu rozdelené na:
Odchýlky v chromozómových súboroch rodičov (dedičné);
- embryo alebo plod boli poškodené vystavením pesticídom, liekom alebo infekciám (teratogénne);
- spoločné ovplyvnenie nenarodeného dieťaťa genetickými a environmentálnymi faktormi, ktoré jednotlivo nemôžu byť príčinou defektu (multifaktoriálne).
Podľa niektorých údajov môže byť znečistenie biosféry tiež príčinou chorôb v 70% prípadov, rozvoj patológií v 60% a smrť detí v 50% prípadov.
Spájajú sa aj vrodené vývojové chyby plodu a následný abnormálny vývoj detí po narodení odborná činnosť- ak človek dlhodobo prežíva emocionálny stres, pôsobenie prachu, vysokej resp nízke teploty, neustále v kontakte s produktmi chemický priemysel alebo soli ťažkých kovov.
Tiež ak budúca mama trpí výraznou obezitou, môže to byť vážnou príčinou abnormalít vo vývoji nervovej trubice plodu. Takéto zmeny na malom tele plodu môžu byť spôsobené nielen nadmernou hmotnosťou tehotnej ženy, ale aj jej prudkým poklesom o skoré dátumy tehotenstva.
malformácie plodu a následné tehotenstvo
Mnohé CM plodu sú liečiteľné. Po narodení, v závislosti od existujúcej anomálie, dieťa absolvuje priebeh nevyhnutných procedúr na jej nápravu alebo liečbu a pokračuje v bežnom živote. V prípade, že CM plodu je nezlučiteľná so životom dieťaťa mimo maternice, tehotenstvo sa preruší. Šesť mesiacov po tomto postupe si môžete naplánovať ďalšie tehotenstvo. Sú chvíle, keď sa páru odporúča počkať jeden rok. Počas tejto doby budúci rodičia absolvujú určité genetické testy a štúdie, podľa ktorých lekár určí, kedy je možné počať dieťa.
Pri príprave na ďalšie tehotenstvo sa pár musí vyhnúť vplyvu negatívnych faktorov, zdravý životný štýlživot, užite multivitamín na posilnenie tela.
16769 0
Vrodené chyby plodu zaujímajú v štruktúre príčin perinatálnej úmrtnosti 2. – 3. miesto a ich frekvencia sa v posledných rokoch výrazne zvyšuje. V tomto ohľade je obzvlášť dôležitá včasná diagnostika malformácií, ktorá prispieva k včasnému vyriešeniu otázky možnosti ďalšieho predĺženia tehotenstva, ktorá je zase určená typom anomálie, kompatibilitou so životom a prognózou pre postnatálne obdobie. rozvoj.
Klasifikácia bežných malformácií centrálnej nervový systém(CNS) môže byť reprezentovaný takto:
1. Hydrocefalus:
a) stenóza cerebrálneho akvaduktu;
b) otvorený hydrocefalus;
c) Dandy-Walkerov syndróm.
2. Papilóm choroidálneho plexu.
3. Poruchy neurálnej trubice:
a) rázštep chrbtice;
b) anencefália;
c) cefalokéla.
4. Mikrocefália.
Hydrocefalus sa vyskytuje s frekvenciou 0,3-0,8 na 1000 živonarodených detí. Vo väčšine prípadov je vrodený hydrocefalus spôsobený obštrukciou v jednej z častí cirkulačnej dráhy cerebrospinálnej tekutiny (CSF). Často sa hydrocefalus kombinuje s inými anomáliami: v 37% prípadov je sprevádzaný ďalšou intrakraniálnou patológiou - hypoplázia corpus callosum, cefalokéla, arteriovenózne anomálie, arachnoidné cysty; extrakraniálne anomálie - v 63%. Medzi nimi malformácie obličiek (jednostranná a bilaterálna agenéza a dysplázia), srdcové chyby (defekt komorového septa, Fallotova tetráda), meningomyelokéla, rozštiepenie hornej pery, tvrdého a mäkkého podnebia, agenéza konečníka a hrubého čreva, gonád dysgenéza, treba upozorniť na Meckeleho syndróm. Chromozomálne abnormality sa nachádzajú u 11 % plodov – trizómia 21 párov, vyvážené translokácie, mozaika.
Hydrocefalus je reprezentovaný tromi hlavnými formami:
- stenóza cerebrálneho akvaduktu;
- otvorený hydrocefalus;
- Dandy-Walkerov syndróm.
Stenóza akvaduktu mozgu(SVM) je forma obštrukčného hydrocefalu spôsobená zúžením Sylviovho akvaduktu. Špecifická frekvencia SVM dosahuje 43 %, pomer detí mužského a ženského pohlavia je 1:8. Anomália má polyetiologický charakter: genetické, infekčné, teratogénne a nádorové faktory, medzi ktorými prevládajú infekčné (50 %). V experimentálnych štúdiách bola potvrdená úloha toxoplazmózy, syfilisu, cytomegalovírusovej infekcie, mumpsu a chrípky.
V určitej časti pozorovaní je príčinou stenózy mozgového akvaduktu genetická patológia, ktorý môže byť zdedený recesívnym typom spojeným s X chromozómom. Dedičnosť viazaná na pohlavie sa považuje za zriedkavú príčinu WMS, pretože sa vyskytuje s frekvenciou 1 prípad na 200 súrodencov probandov s hydrocefalom. Je však možné, že tento typ dedičstva je medzi mužskými deťmi 25 %. Predpokladá sa, že glióm, meningióm, neurofibromatóza a tuberózna skleróza vedú k stenóze akvaduktu v dôsledku kompresného mechanizmu a otvorenému hydrocefalu - v dôsledku edému bielej hmoty a vonkajšej kompresie. Kombinované anomálie sa vyskytujú u 16 % detí.
Diagnóza tejto formy hydrocefalu je založená na detekcii expanzie laterálnych a III. komôr pri nezmenených rozmeroch IV komory ultrazvukom. Je potrebné vykonať dôkladné skenovanie chrbtice plodu, aby sa vylúčili anomálie súvisiace s pohlavím (obr. 1).
Ryža. 1. Tehotenstvo 21 týždňov. Obštrukčný hydrocefalus
Predpoveď: úmrtnosť v detskom veku sa pohybuje od 11-30%; intelektuálny rozvoj môže byť normálne.
Pôrodnícka taktika: pred dosiahnutím životaschopnosti plodu je indikované ukončenie tehotenstva; pri stanovení diagnózy v neskorších štádiách je spôsob pôrodu určený výlučne pôrodníckymi indikáciami.
Otvorený hydrocefalus(OG) - rozšírenie komôr mozgu a jeho subarachnoidálneho systému v dôsledku obštrukcie extraventrikulárneho systému odtokových ciest cerebrospinálnej tekutiny.
Otvorený hydrocefalus je druhým najčastejším, predstavuje 38 % všetkých prípadov hydrocefalu. Etiológia OH nebola objasnená. OH sa zisťuje u detí s defektmi chrbtice a obliteráciou predného sagitálneho sínusu, subarachnoidálnymi hemorágiami, papilómom choroidálneho plexu a absenciou Paccioniho granulácií. Subarachnoidálne krvácanie je najčastejšou príčinou otvoreného hydrocefalu u novorodencov; v prenatálnom období je extrémne zriedkavé. Zriedkavo sa tiež dedí, hoci frekvencia výskytu dosahuje 1 – 2 %, čo je výrazne viac ako v bežnej populácii.
Patogenéza: mechanická obštrukcia extraventrikulárneho systému mozgu a zhoršená reabsorpcia CSF vedú k rozšíreniu subarachnoidálneho priestoru a potom mozgových komôr; vnútorný hydrocefalus sa vyvíja na pozadí obštrukcie cerebrálneho akvaduktu v dôsledku zvýšeného intrakraniálneho tlaku.
Prenatálna diagnostika OH sa vykonáva pomocou dynamického ultrazvukového skenovania. V tomto prípade je patognomickým znakom rozšírenie subarachnoidnej cisterny.
Predpoveď: úmrtnosť dosahuje 11 %. Väčšina prežívajúcich detí si zachováva normálnu inteligenciu. Pri kombinácii OH s defektmi neurálnej trubice alebo papilómom choroidálneho plexu je prognóza nepriaznivejšia.
Pôrodnícka taktika: v skorých štádiách zisťovania OH je indikované ukončenie gravidity, pri donosenej gravidite sa pôrod uskutočňuje prirodzenými pôrodnými cestami.
Pre dandy-walker syndróm Typická je kombinácia nasledujúcich anomálií:
1) hydrocefalus rôzneho stupňa;
2) cysty zadnej lebečnej jamy;
3) defekty cerebelárneho vermis, cez ktoré cysta komunikuje s dutinou IV komory.
Syndróm sa vyskytuje u 12 % detí s vrodeným hydrocefalom. Etiológia nie je známa. Tento syndróm môže byť jedným z prejavov genetických ochorení (Meckelov a Warburgov syndróm) a môže byť detekovaný aj pri chromozomálnych aberáciách (Turnerov syndróm, 6p-, 9gh+, trizómia 9, triploidia). V zriedkavých prípadoch je možná autozomálne recesívna dedičnosť s rizikom recidívy až 25%.
Patoembryogenéza. Podľa Dandy-Walkerovej teórie atrézia dier Luschka a Magendie zvyčajne vedie k expanzii komorového systému. Syndróm je komplexná anomália vo vývoji stredných štruktúr mozgu v oblasti kosoštvorcovej jamky. Hypoplázia cerebelárneho vermis a cysty zadnej jamy sa vyskytujú sekundárne v dôsledku kompresie jej ostro rozšírenej IV komory. Existuje tiež nerovnováha v produkcii cerebrospinálnej tekutiny v laterálnych, III a IV komorách. Defekt mozgového červa sa mení od úplnej aplázie po mierne štiepenie. Napriek tomu, že hydrocefalus je hlavným diagnostickým znakom Dandy-Walkerovho syndrómu, väčšina detí ho nemá v čase narodenia, prejavuje sa však v prvých mesiacoch života dieťaťa.
Syndróm je často (vo viac ako 50% prípadov) kombinovaný s inými malformáciami centrálneho nervového systému (agenéza corpus callosum, encefalokéla), malformáciami obličiek (polycystické) a srdca (defekt komorového septa).
Diagnostika: táto anomália je indikovaná detekciou cystických útvarov v zadnej lebečnej jamke počas echografie; patognomickým akustickým znakom syndrómu je defekt mozgového červa, cez ktorý cysta komunikuje so štvrtou komorou.
Predpoveď nepriaznivé: úmrtnosť dosahuje 50%, 50-60% prežívajúcich detí má zaostávanie v intelektuálnom vývoji.
Pôrodnícka taktika: potratu kedykoľvek.
Papilóm choroidálneho plexu(PSS). Intrakraniálny novotvar sa vyskytuje s frekvenciou 0,6% všetkých nádorov mozgu zistených u dospelých a 3% u detí. Papilóm môže byť lokalizovaný v ktorejkoľvek časti komorového systému, ale častejšie sa nachádza na úrovni vestibulu bočných komôr. Charakteristická je jednostranná lokalizácia nádoru, aj keď nie je vylúčený obojstranný proces. Najčastejšie je PSS reprezentovaný tkanivom klkov, histologicky podobným tkanivu intaktného choroidálneho plexu a je benígny. Je však možná malignita nádoru s klíčením do susedného nervového tkaniva. Papilóm choroidálneho plexu je zvyčajne kombinovaný s hydrocefalom.
Etiológia neznámy. Vo svetovej praxi sú známe izolované pozorovania PSS u pacientov s Aicardiho syndrómom (toto ochorenie je viazané na X chromozóm a je charakterizované agenézou corpus callosum, chorioretinálnymi lakúnami, miechovými anomáliami, epilepsiou a mentálnou retardáciou).
PSS je diagnostikovaná u detí s hydrocefalom počas neurosonografie alebo röntgenu. V detstve by sa metóda výberu mala považovať za kontrast Počítačová tomografia. Ultrazvukové vyšetrenie je najinformatívnejšou metódou prenatálnej diagnostiky. Echografickými kritériami PSS sú asymetria tvaru a veľkosti kontralaterálnych komôr, vizualizácia slabo echogénnych útvarov priliehajúcich k normálnemu plexus choroideus vo vestibule laterálnych komôr. Prenatálne sa PSS III a IV komôr nezistí.
Metódou voľby terapie PSS je chirurgické odstránenie nádorov. Pri benígnej povahe procesu môže mať chirurgická liečba priaznivý výsledok, ale operácia je technicky zložitá a je sprevádzaná veľkou stratou krvi. Pri malígnej lézii (vo viac ako 20% prípadov) je prognóza nepriaznivá. Úmrtnosť na PSS dosahuje 35 % a 72 % prežívajúcich detí má rôzne stupne závažnosti defektov v mentálnom a duševnom vývoji.
Pôrodnícka taktika: pôrod vedie cez prirodzené pôrodné cesty; použitie operácií vaginálneho pôrodu je kontraindikované. Pôrod sa odporúča vo veľkých perinatologických centrách, kde je možné poskytnúť urgentnú neonatologickú a detskú neurochirurgickú starostlivosť. Otázka použitia cisárskeho rezu na zníženie rizika pôrodnej traumy s intrakraniálnym krvácaním nie je definitívne vyriešená.
Vybrané prednášky z pôrodníctva a gynekológie
Ed. A.N. Strizhakova, A.I. Davydová, L.D. Belotserkovtseva
Relevantnosť. Prenatálna diagnostika (PD) vrodených vývojových chýb plodu je mimoriadne dôležitou zložkou prenatálnej starostlivosti, ktorá umožňuje predchádzať narodeniu detí s ťažkými neopraviteľnými vývojovými chybami, so spoločensky významnými smrteľnými génovými a chromozomálnymi ochoreniami. Toto je hlavný faktor pri znižovaní chorobnosti, invalidity a úmrtnosti. Okrem toho prenatálna detekcia vrodených anomálií mozgu u plodu ovplyvňuje pôrodnícku taktiku a znižuje negatívne psychologické a sociálne dôsledky pre matku a pre rodinu ako celok, keďže vrodené vývojové chyby (VM) mozgu sú najmä ťažké a nevyliečiteľné choroby. (včasná a kvalitná diagnostika malformácií CNS dokáže objektívne predpovedať situáciu a prispieť k správna voľba taktika liečby). Kompetentná prenatálna diagnostika v kombinácii so zberom kompletných informácií zameraných na identifikáciu rôznych rizikových faktorov teda umožňuje lekárovi a rizikovej tehotnej adekvátne pristupovať k problematike pôrodu.
Metódy, slúžiace na prenatálnu (prenatálnu) diagnostiku, je vhodné rozdeliť na nepriamu, kedy je objektom štúdie tehotná žena, a priamu, kedy sa vyšetruje samotný plod. Ten môže byť invazívny a neinvazívny.
Nepriame metódy zahŕňajú štúdium pôrodníckej a gynekologickej histórie, lekárske genetické poradenstvo, ako aj bakteriologické a sérologické štúdie. Samostatne je potrebné povedať o vykonávaní biochemických skríningových testov zameraných na hodnotenie hladín fetoproteínu, estriolu, ľudského chorionického gonadotropínu atď. Je potrebné poznamenať, že hlavným účelom nepriamych metód je výber žien z vysoko rizikových skupín pre ďalšie hĺbkové monitorovanie už na úrovni centier reprodukčného zdravia. Hlavné indikácie na odoslanie tehotnej ženy na PD sú približne rovnaké na celom svete. Tie obsahujú:
[1
] vek ženy nad 35 rokov;
[2
] prítomnosť aspoň dvoch spontánne potraty(potraty) na začiatku tehotenstva;
[3
] prítomnosť v rodine dieťaťa alebo plodu z predchádzajúceho tehotenstva s Downovou chorobou, inými chromozomálnymi chorobami, s mnohopočetnými vrodenými chybami, rodinným prenášaním chromozomálnych prestavieb;
[4
] mnohé monogénne choroby predtým diagnostikované v rodine alebo blízkych príbuzných;
[5
] užívanie viacerých farmakologických prípravkov pred a v počiatočných štádiách tehotenstva;
[6
] prekonané vírusové infekcie (hepatitída, rubeola, toxoplazmóza atď.);
[7
] ožiarenie jedného z manželov pred počatím.
prečítajte si aj článok: Toxoplazmóza(na webovú stránku)
Hlavné neinvazívne metódy sú [ 1 ] ultrazvukové (sonografické) vyšetrenie (ultrazvuk) a [ 2 ] zobrazovanie magnetickou rezonanciou (MRI).
Ultrazvukové vyšetrenie CNS a miechy u plodu je jednou z najdôležitejších a najzodpovednejších úloh prenatálnej echografie, pretože má významný vplyv na optimalizáciu pôrodníckej taktiky a rozhodnutie rodičov o predĺžení alebo ukončení tehotenstva. Optimálne načasovanie echografického vyšetrenia plodu na vylúčenie väčšiny defektov a ochorení centrálneho nervového systému je:
[1
] 11. - 14. týždeň tehotenstva;
[2
] 19. - 21. týždeň tehotenstva;
[3
] 30 - 33 týždňov tehotenstva.
To zodpovedá počiatočným štádiám prejavov rôznych skupín malformácií a ochorení centrálneho nervového systému a tiež zabezpečuje kontinuitu diagnostiky a všeobecne uznávaných štandardov vedenia tehotenstva a pôrodu. Súčasne by schéma ultrazvukového vyšetrenia plodu mala zahŕňať posúdenie echografickej anatómie plodu so štúdiom kostí lebečnej klenby, hlavných štruktúr mozgu, golierového priestoru, profilu, očníc, nosa. kosti, orientácia srdca, chrbtice, prednej brušnej steny, žalúdka, čriev, močového mechúra a končatín.
Údaje získané množstvom autorov svedčia o vysokej diagnostickej hodnote ultrazvuku v prenatálnej diagnostike vrodených vývojových chýb, ktorá umožňuje identifikovať viac ako 80–90 % vývojových chýb plodu počas tehotenstva. Významnú časť hrubej patológie vývoja plodu je možné zistiť v prvom trimestri, v závislosti od načasovania vyšetrenia a všetkých požiadaviek skríningového protokolu na posúdenie anatómie plodu.
Použitie moderných ultrazvukových prístrojov expertnej triedy, transvaginálna echografia využívajúca 3/4D režimy výrazne zvyšuje presnosť diagnostiky. Štúdium mozgu plodu v strednej sagitálnej rovine, ktoré sa vo väčšine prípadov stalo prakticky možným pomocou trojrozmernej echografie, umožňuje diferencovaný prístup k hodnoteniu normy a patológie stredových mozgových štruktúr. Axiálne rezy mozgu plodu používané v rutinnej ultrazvukovej praxi neumožňovali získať jasný obraz corpus callosum. Rozšírenie protokolu prvej skríningovej štúdie o hodnotenie „intrakraniálnej transparentnosti“ zvýšilo presnosť diagnostiky spina bifida v prvom trimestri gravidity.
Využitie farebného dopplerovského mapovania na vizualizáciu mozgových ciev u plodu v ranej ontogenéze spoľahlivo umožňuje vizualizáciu cievy zásobujúcej špecifickú štruktúru o 2–5 týždňov skôr ako ich štandardná echografická detekcia už na konci 1. a na začiatku r. 2. trimester tehotenstva. Komplexné hodnotenie vývoja sulciových a subarachnoidálnych priestorov umožňuje diagnostikovať porušenie tvorby mozgovej kôry už v druhom trimestri tehotenstva. Anomálie vo vývoji centrálneho nervového systému, ako je narušenie embryogenézy a skorá organogenéza plodu, sa dajú zistiť pomocou modernej echografie až do 21. týždňa tehotenstva. Zároveň echografické znaky deštruktívnych lézií a objemových útvarov CNS možno diagnostikovať iba v II a III trimestri tehotenstva (S.M. Voevodin, 2012).
Transvaginálna echografia so štúdiom anatomických štruktúr plodu pomocou ultrazvuku v 11-14 týždni tehotenstva je vysoko informatívna metóda prenatálnej diagnostiky na začiatku tehotenstva, ktorá umožňuje odhaliť viac ako polovicu všetkých vrodených malformácií, ktoré sú hrubé väčšinou.
Je tiež potrebné poznamenať hodnotu pre diagnostiku vrodených malformácií takého ultrazvukového znaku, ako je polyhydramnios. Závažnosť polyhydramniónu koreluje s frekvenciou vrodených malformácií plodu. Bol vytvorený priamy vzťah medzi plodová voda a výskyt vrodených vývojových chýb plodu.
Vo všeobecnosti úspešnosť diagnostiky závisí aj od typu vrodených malformácií (niekoľko anomálií je dosť ťažké diagnostikovať), gestačného veku, v ktorom sa štúdia vykonáva, množstva plodovej vody a ústavných charakteristík pacienta. (ťažká obezita spôsobuje ťažkosti pri transabdominálnom skenovaní). Ďalšími faktormi, ktoré sťažujú správnu interpretáciu výsledkov skríningu, môže byť gestačný vek, viacpočetné tehotenstvo, etnicita rodičov, cukrovka u matky. Prenatálna diagnostika agenézy corpus callosum (ktorého hlavnými podmienkami detekcie sú II-III trimestre tehotenstva), spinálna hernia je pomerne komplikovaná. Zároveň, napriek vysokej presnosti a relatívnej jednoduchosti ultrazvukovej diagnostiky takých absolútne smrteľných defektov, ako je akrania a anencefália, niektoré z nich sú detekované po 24 týždňoch tehotenstva, čo môže naznačovať nedostatočne vysokú kvalifikáciu lekárov, nedodržiavanie skríningu termíny a vyšetrovacie metódy., neskoré odporúčanie žien na druhý alebo tretí stupeň vyšetrenia. Ďalším negatívnym faktorom môže byť odmietnutie prenatálnej karyotypizácie ženami. Okrem toho také otázky prenatálnej ultrazvukovej diagnostiky vrodených vývojových chýb centrálneho nervového systému ako odlišná diagnóza so zriedkavými poruchami.
MRI je vysoko informatívna metóda prenatálnej diagnostiky a možno ju použiť pri podozrení na malformácie plodu pri ultrazvukovom vyšetrení, najmä v prípadoch anomálií CNS. V II a III trimestri gravidity použitie MRI ako dodatočného vyšetrenia zlepšuje diagnostiku defektov CNS a umožňuje spresniť diagnózu v prípade patológie mozgovej kôry, stredových štruktúr mozgu, zadnej lebečnej jamy, a poruchy dynamiky CSF. MRI možno úspešne použiť v prípadoch, keď výsledky ultrazvuku nie sú dostatočne informatívne. Zatiaľ čo MRI je schopná potvrdiť diagnózu v USA a poskytnúť podrobnejšie údaje, relatívne vysoká cena, nedostatok štandardizovaných referenčných hodnôt a obmedzená dostupnosť MRI sú dôvodmi, prečo US zostáva štúdiou voľby na diagnostiku CM plodu.
prečítajte si tiežčlánok "MRI mozgu plodu: prehľad" od S. Yazbeka a P.E. Grant. Neurografia, ročník 5, číslo 5, 1. septembra 2015, s. 181-191(11) [Časť č. 1] a [Časť č. 2]
O Je MRI bezpečná počas tehotenstva? môžeš čítať
Štúdium markerových embryonálnych proteínov v krvnom sére matky. V posledných rokoch zohráva obzvlášť dôležitú úlohu štúdium markerových embryonálnych proteínov v krvnom sére matky, ako je alfa-fetoproteín (AFP), chorionický gonadotropín (hCG), voľný estradiol a niektoré ďalšie. Účelom takýchto štúdií je identifikovať ženy s vysokým rizikom, že budú mať deti s vrodenými a dedičnými chybami. Zmeny sérových markerov sú typické pre 90,9 % žien s vrodenými chybami centrálneho nervového systému. Štúdia realizovaná v optimálnom čase (15-16 týždňov tehotenstva) pomocou troch testovacích systémov umožňuje identifikovať až 80 % plodov s poruchami vývoja vnútorných orgánov a až 65 % s chromozomálnymi ochoreniami.
Prenatálna laboratórna diagnostika anomálií neurálnej trubice je založená predovšetkým na stanovení hladiny fetálneho AFP. Tento proteín je hlavnou zložkou fetálneho sérového proteínového systému a stanovuje sa na 30. deň tehotenstva. Zvýšenie hladiny AFP v plodovej vode je znakom otvoreného defektu neurálnej trubice. V druhom trimestri tehotenstva dokáže ultrazvuk spoľahlivo odhaliť abnormalitu mozgu plodu. Keďže sa na tvorbe estriolu podieľa plod aj placenta, hladina estriolu môže slúžiť ako ideálny indikátor fungovania fetoplacentárneho systému. Čím nižšia je hladina hormónu, tým vyššia je pravdepodobnosť vzniku patologického stavu plodu.
Interpretácia izolovaných výsledkov biochemického testovania môže byť zároveň náročná. Pri zvažovaní distribučných kriviek hodnôt hlavných markerov existuje veľká zóna prekrytia medzi normou a patológiou, čo neumožňuje použiť iba jeden marker, je potrebný ich úplný komplex: hCG, AFP a estriol.
Biochemické vyšetrenie v prvom trimestri gravidity, ktoré zahŕňa [ 1 ] stanovenie koncentrácií progesterónu, [ 2 ] nekonjugovaný estriol, [ 3 ] β-frakcia ľudského chorionického gonadotropínu (β-hCG) a [ 4 ] bielkovín spojených s tehotenstvom (7 - 8 alebo 11 - 12 týždňov) je viac efektívna metóda prenatálny skríning ako tradičný „trojitý“ test druhého trimestra, t.j. AFP, β-hCG, estriol - v 16. - 17. týždni tehotenstva.
Napriek pomerne vysokej účinnosti moderných neinvazívnych techník, dostatočne úplné informácie o karyotype embrya, biochemické a genotypové vlastnosti jeho buniek možno získať len na základe vhodných štúdií tkanív samotného plodu alebo jeho provizórneho orgány (placenta, chorion) získané invazívne kedykoľvek počas tehotenstva. Amniocentéza sa najčastejšie vykonáva na zistenie chromozomálnych abnormalít a genetických mutácií, ale plodová voda sa môže použiť aj na diagnostiku defektov neurálnej trubice. Najčastejšie možné riziká postupmi sú spontánny potrat (0,5 % až 1,0 %), špinenie po zákroku, infekcia, pretrhnutie membrán a poranenie alebo strata plodu.
Najinformatívnejšie je komplexné vyšetrenie tehotných žien pomocou moderných ultrazvukových technológií v kombinácii s biochemickým skríningom, ktoré zvyšuje presnosť diagnostiky vrodených vývojových chýb centrálneho nervového systému plodu vo včasných štádiách druhého trimestra. Okrem toho detekcia príznakov hypoxie plodu môže naznačovať možnú prítomnosť nielen fetoplacentárnej insuficiencie, ale aj vrodenej patológie, pretože hypoxia a intrauterinná retardácia rastu výrazne zhoršujú prognózu CM zlučiteľnú so životom.
Malo by sa pamätať, Čo vrodené anomálie mozgu, zistené prenatálne alebo v novorodeneckom období, nepočítajúc netrvalé vonkajšie malformácie, [ !!! ] NEMUSÍ mať žiadne špecifické klinické príznaky:
[1 ] pri neurosonografických a magnetických rezonančných štúdiách treba okrem špecifických nozologických foriem vrodených vývojových chýb mozgu (Chiariho syndróm, Dandy-Walkerov syndróm, okluzívny hydrocefalus atď.) venovať pozornosť hypoplázii, ktorá je oveľa bežnejšia;
[2 ] pri absencii prenatálnej a novorodeneckej diagnostiky pomocou ultrazvuku a MRI sa objavujú podmienky na neskorú diagnostiku v období, keď sú neuropsychiatrické symptómy na prvom mieste v klinickom obraze, pričom základom pre vznik takýchto ochorení môže byť prítomnosť mentálnej retardácie, intrakraniálnej hypertenzie. diagnózy ako detská mozgová obrna, hydrocefalus atď.
prečítajte si aj článok: Neurosonografia: Ultrazvuková diagnostika perinatálnych lézií CNS(na laesus-de-liro.live-journal.com)
prečítajte si aj článok: syndróm Dandyho Walkera(na webovú stránku)
prečítajte si aj článok: Anomália (malformácia) Chiari typ I(na webovú stránku)
prečítajte si aj článok: Mozgová obrna(na webovú stránku)
Najvyššia výpovedná hodnota konvulzívneho syndrómu ukazuje na prítomnosť agenézy corpus callosum alebo agýrie, hypertenzno-hydrocefalický syndróm na prítomnosť vrodeného hydrocefalu a syndróm motorických porúch na prítomnosť hernie chrbtice.
Záver. Na zabránenie narodenia detí s poruchami centrálneho nervového systému (neurálnej trubice) sa v súčasnosti používajú tieto metódy: [ 1 ] primárny skríningový test na zistenie štrukturálnych abnormalít plodu vrátane otvoreného/uzavretého defektu neurálnej trubice (anencefália, encefalokéla, spina bifida) – ultrazvuk plodu v druhom trimestri tehotenstva; [ 2 ] stanovenie hladín AFP v sére matky; [ 3 ] genetické poradenstvo prípadov s pozitívnymi výsledkami skríningu v čase defektov neurálnej trubice (ultrazvuk + AFP v sére matky); [ 4 ] prenatálna magnetická rezonancia ako dodatočná metóda zobrazovania plodu; [ 5 ] diagnostická amniocentéza na posúdenie karyotypu plodu, stanovenie hladiny AFP v plodovej vode a aktivity acetylcholínesterázy; [ 6 ] pri potvrdení prítomnosti defektov neurálnej trubice u plodu by mali byť rodine ponúknuté možnosti zvládnutia skutočného tehotenstva - ako predĺženie s možnosťou pred- alebo postnatálnej korekcie defektu, tak aj ukončenie tehotenstva s defektom nezlučiteľným so životom ; spôsob pôrodu sa volí individuálne - môže to byť prirodzený (vaginálny) pôrod (pri absencii kontraindikácií) s monitorovaním srdcovej frekvencie plodu a pôrod cisárskym rezom; [ 7 ] popôrodné konzultácie na informovanie ženy o príčine vzniku defektu neurálnej trubice u plodu, možnosti recidívy takéhoto stavu u plodu pri ďalších tehotenstvách a prevencii, najmä odporúčanie užívať kyselinu listovú na 5000 mcg denne rodičmi 3 mesiace pred počatím a počas tehotenstva v prvom trimestri.