Kongenitalne malformacije fetusa zauzimaju 2-3 mjesto u strukturi uzroka perinatalne smrti fetusa i novorođenčeta. Od velike važnosti rana dijagnoza malformacije, što je potrebno za pravovremeno rješavanje pitanja mogućnosti produljenja trudnoće, što je određeno vrstom defekta, kompatibilnošću sa životom i prognozom za postnatalni razvoj. Ovisno o etiologiji, razlikuju se nasljedne (genetske), egzogene i multifaktorijalne kongenitalne malformacije fetusa. Nasljedne uključuju malformacije nastale mutacijama, tj. trajne promjene u nasljednim strukturama u gametama ili zigoti. Ovisno o razini na kojoj je mutacija nastala (geni ili kromosomi), razlikuju se monogeni sindromi i kromosomske bolesti. Egzogeni uključuju nedostatke uzrokovane štetnim djelovanjem egzogenih čimbenika. Ovi čimbenici, djelujući u razdoblju gametogeneze ili trudnoće, dovode do pojave kongenitalnih malformacija bez poremećaja strukture nasljednog aparata.
Malformacije multifaktorijalnog podrijetla nazivaju se defekti koji su nastali pod kombiniranim utjecajem genetskih i egzogenih čimbenika. Također postoje izolirani (lokalizirani u jednom organu), sistemski (unutar jednog organskog sustava) i multipli (u organima dva ili više sustava) defekti.
GREŠKE SREDIŠNJEG ŽIVČANOG SUSTAVA
Klasifikacija najčešćih malformacija središnjeg živčanog sustava:
1. Hidrocefalus:
Stenoza cerebralnog akvadukta;
Otvoreni hidrocefalus;
Dandy-Walkerov sindrom.
2. Papiloma koroidnog pleksusa.
3. Defekti neuralne cijevi:
- spina bifida;
anencefalija;
Cefalokela.
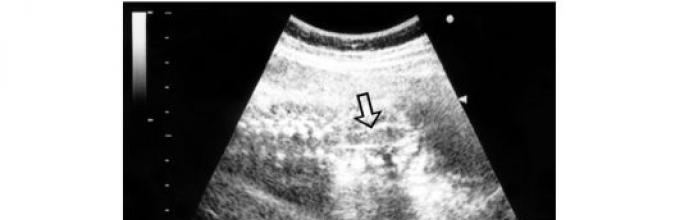
4. Mikrocefalija. Hidrocefalus
Hidrocefalus- povećanje veličine ventrikula mozga s istodobnim povećanjem intrakranijalnog tlaka, popraćeno u većini slučajeva povećanjem veličine glave (slika 28).
Riža. 28. Sonografska slika teškog fetalnog hidrocefalusa (strelice označavaju oštro proširene ventrikule mozga, čija je kora značajno istanjena, veličina glave fetusa prelazi normalne vrijednosti za određenu gestacijsku dob)
Ventrikulomegalija je izolirano povećanje veličine ventrikula, koje nije popraćeno povećanjem veličine glave. Hidrocefalus se opaža s učestalošću od 0,1-2,5 na 1000 novorođenčadi. Oko 60% fetusa s hidrocefalusom su dječaci. Hidrocefalus može biti posljedica mnogih bolesti različite etiologije. U većini slučajeva razvija se kao posljedica kršenja odljeva cerebrospinalne tekućine. Komunikantni oblik hidrocefalusa uzrokovan je ekstraventrikularnim
kularna opstrukcija, dok je opstruktivni oblik intraventrikularna opstrukcija. Povremeno hidrocefalus dovodi do povećane proizvodnje cerebrospinalne tekućine (na primjer, na pozadini papiloma vaskularnih pleksusa) ili kršenja njegove reapsorpcije u subarahnoidnom prostoru.
Ekstrakranijalne anomalije kod hidrocefalusa javljaju se u 63% slučajeva: agenezija i displazija bubrega, defekt ventrikularnog septuma, Fallot tetralogija, meningomijelokela, splitting Gornja usna, meko i tvrdo nepce, atrezija anusa i rektuma, disgeneza gonada. Hidrocefalus je predstavljen uglavnom stenozom cerebralnog akvadukta (suženje Silvijevog akvadukta); otvoreni hidrocefalus (proširenje ventrikula mozga i subarahnoidnog sustava mozga kao rezultat opstrukcije ekstraventrikularnog sustava izlaznih puteva cerebrospinalne tekućine); Dandy-Walkerov sindrom (kombinacija hidrocefalusa, ciste stražnje lubanjske jame, defekata u vermisu malog mozga, kroz koje cista komunicira sa šupljinom IV ventrikula). Kada se otkrije hidrocefalus, potrebno je pažljivo procijeniti anatomiju moždanih struktura, kao i kralježnice, kako bi se isključila spina bifida. Sveobuhvatni pregled fetusa trebao bi uključivati ehokardiografski pregled, budući da se hidrocefalus često kombinira s prirođenim srčanim manama. Uz hidrocefalus prije razdoblja održivosti fetusa, preporučljivo je razgovarati o pitanju prekida trudnoće s roditeljima. Kod produljenja trudnoće prikazuje se dinamički ultrazvučni nadzor svaka 2 tjedna. S porastom hidrocefalusa nakon dostizanja zrelosti pluća fetusa, treba postaviti pitanje ranog poroda i ranžiranja. Učinkovitost prenatalne ventrikularne premosnice još nije dokazana i ova operacija nema široku primjenu. Carski rez indiciran je samo u slučaju teške makrocefalije i odsutnosti drugih malformacija. U prisutnosti velikih kombiniranih anomalija koje pogoršavaju prognozu života, operacija izbora je cefalocenteza.
Defekti neuralne cijevi. Ovaj pojam uključuje anencefaliju, cefalokelu i spina bifida.
spina bifida- anomalija u razvoju kralježnice, koja je posljedica kršenja procesa zatvaranja neuralne cijevi (slika 29).
Izlaz kroz defekt u ovojnicama leđne moždine naziva se meningokela. Ako hernialna vrećica sadrži živčano tkivo, formacija
 Riža. 29. Sonografska slika spina bifida u lumbosakralnoj kralježnici (označeno strelicom)
Riža. 29. Sonografska slika spina bifida u lumbosakralnoj kralježnici (označeno strelicom)
naziva se meningomijelokela. razlikovati spina bifida cystica(cistični oblik kile kralježnice s formiranjem hernijalne vrećice koja sadrži membrane mozga i/ili supstancu mozga) i spina bifida occulta(skriveni oblik, koji nije popraćen stvaranjem hernialne izbočine). Najčešće je ovaj nedostatak lokaliziran u lumbalnoj i sakralnoj kralježnici. Učestalost pojavljivanja spina bifida ovisi o geografskoj regiji. U nekim područjima Ujedinjenog Kraljevstva učestalost ove mane je 4 na 1000 novorođenčadi. U Sjedinjenim Državama ta je brojka 0,5 na 1000, iako ovisi o rasnim i geografskim karakteristikama. spina bifida- malformacija koja se javlja zbog kršenja zatvaranja neuralne cijevi u 4. tjednu embrionalnog razvoja. Ova se anomalija nasljeđuje na višefaktorski način. spina bifida a može nastati kao posljedica hipertermije majke, ako ima dijabetes melitus, izloženosti teratogenim čimbenicima, a također biti dio genetskih sindroma (s izoliranim mutiranim genom) ili kromosomskih abnormalnosti (trisomije 13 i 18 parova kromosoma, triploidija, neuravnotežena translokacija ili prstenasti kromosom). Spinalna kila povezana je s više
više od 40 sindroma višestrukih malformacija (hidrocefalus, urođene srčane mane i genitourinarni sustav).
Prenatalni pregled uključuje određivanje kariotipa i detaljan ultrazvučni pregled. Posebna pažnja treba posvetiti anatomiji glave, srca, ruku i nogu. Ako se meningomijelokela otkrije prije vitalnosti fetusa, ženi treba ponuditi medicinski prekid trudnoće. Kod produljenja trudnoće indiciran je dinamički ultrazvuk svaka 2-3 tjedna za procjenu pojave drugih znakova (na primjer, ventrikulomegalija). Roditeljima je potrebna konzultacija s neurokirurgom kako bi se razmotrile mogućnosti kirurške intervencije nakon poroda (zatvaranje defekta ili premosnice), kao i prognoza za život i zdravlje djeteta. Porođaj treba provesti u velikim perinatalnim centrima čim pluća fetusa dostignu dovoljnu zrelost. Empirijski rizik od recidiva spina bibida iznosi 3-5%. Primjena velikih doza folne kiseline (4 mg), započeta 3 mjeseca prije planirane trudnoće i nastavljena tijekom prve polovice trudnoće, može značajno smanjiti rizik od defekta.
Svaki otvoreni defekt neuralne cijevi treba zatvoriti unutar prva 24 sata života. Antibakterijska terapija započeta odmah nakon rođenja može smanjiti rizik od zaraznih komplikacija. Prognoza za život i zdravlje ovisi o razini lokacije meningomijelokele, kao io broju i prirodi povezanih anomalija. mentalni razvoj djeca koja pri rođenju imaju normalan opseg glave i pravilno formiran mozak ne pate. Bolesnici s meningomijelokelom smještenom na L2 i iznad gotovo su uvijek prisiljeni koristiti invalidska kolica.
Anencefalija(pseudocefalija, ekstrakranijalna disencefalija) - odsutnost hemisfera velikog mozga i većeg dijela svoda lubanje, dok postoji defekt frontalne kosti iznad supraorbitalne regije, nema temporalne i dijela okcipitalne kosti. Gornji dio glave prekriven je vaskularnom membranom. Strukture srednjeg mozga i diencefalona su djelomično ili potpuno uništene. Hipofiza i romboidna jamica su uglavnom očuvane. Tipične manifestacije uključuju izbuljene oči, velik jezik i kratak vrat. Ova se patologija javlja s učestalošću od 1 u 1000. Češće ga
nalaze se kod novorođenih djevojčica. Acrania(egzencefalija) - odsutnost svoda lubanje u prisutnosti fragmenta moždanog tkiva. To je rjeđa patologija od anencefalije. Anencefalija je rezultat neuspjeha zatvaranja rostralne neuropore unutar 28 dana od oplodnje. Zapažene su multifaktorijalno i autosomno recesivno nasljeđivanje, kromosomske abnormalnosti. Čimbenici rizika uključuju dijabetes majke. U pokusima na životinjama utvrđena je teratogenost zračenja, salicilata, sulfonamida i povišene razine ugljičnog dioksida. Sonografska dijagnoza se može postaviti već u 12-13 tjednu trudnoće. Anencefalija i akranija su apsolutno smrtonosne malformacije, stoga u oba slučaja ženi treba ponuditi prekid trudnoće. Sva novorođenčad s anencefalijom i akranijom umiru unutar 2 tjedna od rođenja. Empirijski rizik od ponovne pojave anencefalije je 3-5%. Primjena velikih doza folne kiseline (4 mg), započeta 3 mjeseca prije planirane trudnoće i nastavljena tijekom prve polovice trudnoće, može značajno smanjiti rizik od defekta.
Cefalokela(encefalokela, kranijalna ili okcipitalna meningokela, rascjep lubanje) - izbočenje sadržaja lubanje kroz defekt kosti. Izraz "kranijalna meningokela" odnosi se na izbočenje samo kroz defekt meningealnih membrana. Kada je moždano tkivo u hernijalnoj vrećici, koristi se izraz "encefalokela". Cefalokela je rijetka (1:2000 živorođene djece) i sastavni je dio mnogih genetskih (Meckeleovi sindromi, srednje rascjep lica) i negenetskih (amnionska konstrikcija) sindroma. Cefalokela nastaje kao posljedica nezatvorenosti defekta neuralne cijevi i javlja se u 4. tjednu razvoja. Defekt u lubanji, kroz koji mogu prolabirati membrane mozga i moždanog tkiva, nastaje kao rezultat neodvajanja površinskog ektoderma i neuroektoderma koji leži ispod. Ako se otkrije cefalokela, ženi treba ponuditi medicinski prekid trudnoće. Kod produljenja trudnoće, taktika poroda ovisi o veličini i sadržaju hernialne vrećice. Na velike veličine defekt, prolaps značajne količine moždanog tkiva, kao iu prisutnosti mikrocefalije i hidrocefalusa, prognoza za život i zdravlje je izuzetno nepovoljna.
Dostava po operaciji carski rez nije prikazano u ovim opažanjima. Moguće je preporučiti dekompresiju hernialne vrećice kako bi se stvorili uvjeti za isporuku kroz prirodni rodni kanal. Carski rez se može preporučiti ako postoji mali defekt i ako je kilna vreća mala.
Mikrocefalija (mikroencefalija) je klinički sindrom karakteriziran smanjenjem opsega glave i mentalnom retardacijom. Javlja se s učestalošću od 1 na 1360 novorođenčadi, s kombiniranim anomalijama od 1,6:1000 živorođene djece. Mikrocefalija je polietiološka bolest u čijem nastanku važnu ulogu imaju genetski (kromosomske aberacije, monogeni defekti) i okolišni čimbenici. Prognoza ovisi o prisutnosti pridruženih anomalija. Trisomija na kromosomima 13, 18, Meckelov sindrom su fatalne lezije. Prenatalni pregled treba uključivati određivanje fetalnog kariotipa i temeljit ultrazvučni pregled. U nedostatku popratnih anomalija, prognoza ovisi o veličini glave: što je manja, to je niži indeks intelektualnog razvoja. Mikrocefalija je neizlječiva bolest. Opstetrička taktika - prekid trudnoće prije nego što fetus postigne održivost.
ANOMALIJE STRUKTURA LICA I VRATA
Rascjep lica(rascjep usne i nepca) je linearni defekt koji se proteže od ruba usne do nosnog otvora.
Rascjep nepca, u kombinaciji s rascjepom usne, kroz alveolarne nastavke i tvrdo nepce može se proširiti na nosnu šupljinu ili čak na dno orbite. Bilateralni rascjep usne opažen je u 20%, rascjep usne i nepca - 25%. S jednostranom lezijom, rascjep se češće nalazi lijevo. Rascjep lica čini oko 13% svih malformacija i bilježi se s učestalošću od 1:800 živorođene djece. Dječaci imaju rascjepe češće od djevojčica. Kombinirane anomalije nalaze se u 50% slučajeva s izoliranim rascjepom nepca i samo u 13% s rascjepom usne i nepca. Strukture lica formiraju se između 4. i 10. tjedna trudnoće. Neparne frontonazalne strukture spajaju se s parnim strukturama maksilarne i mandibularne čeljusti.
moje kvržice. U onim promatranjima kada proces fuzije nije dovršen, formiraju se pukotine. U pravilu je moguće dijagnosticirati rascjep lica samo u drugom tromjesečju trudnoće ultrazvučnim probirom. Prenatalno otkrivanje defekta ehografijom je teško, no zahvaljujući ultrazvučnom skeniranju i color doppler kartiranju, mogućnosti njegove dijagnoze se proširuju. Doppler ultrazvuk može vizualizirati kretanje tekućine kroz nos, usta i ždrijelo. U prisutnosti rascjepa, priroda kretanja tekućine se mijenja. Trodimenzionalna ehografija može razjasniti dijagnozu u onim opažanjima kada se u dvodimenzionalnoj studiji sumnjalo na rascjep, ali nije dobivena njegova jasna vizualizacija. Moguće je dijagnosticirati anomaliju uz pomoć fetoskopije, uključujući embrioskopiju. U nedostatku povezanih anomalija koriste se općeprihvaćene opstetričke taktike, bez obzira na vrijeme dijagnoze. Uzimanje folne kiseline prije i tijekom sljedeće trudnoće može smanjiti rizik od rascjepa.
Rascjep gornje usne (rascjep) ne ometa čin sisanja i samo je kozmetički nedostatak. S kombinacijom rascjepa gornje usne, čeljusti i tvrdog nepca (rascjep nepca) bilježe se funkcionalni poremećaji: pri sisanju mlijeko istječe kroz nos zbog komunikacije s usnom šupljinom; mlijeko se može udisati. Prognoza je povoljna: moderne kirurške metode omogućuju postizanje korekcije kozmetičkih i funkcionalnih nedostataka.
cistični higrom(limfangiom ili posljedica začepljenja jugularnog limfnog stabla) je encistična nakupina tekućine (slika 30). Karakterizira ga prisutnost pojedinačnih ili višestrukih cista mekog tkiva u vratu, koje su posljedica poremećaja u limfnom sustavu. Cistični higromi javljaju se s učestalošću od 1:200 spontanih pobačaja (kokcyx-parijetalna veličina fetusa preko 30 mm). Cistični higrom često se kombinira s kromosomskim aberacijama (Turnerov sindrom, trisomija 13, 18, 21 para kromosoma, mozaicizam). Kao izolirana anomalija, nasljeđuje se autosomno recesivno. Prognoza: u većini slučajeva plod ugine u prva dva tromjesečja trudnoće. Oko 90% zahtijeva kirurško liječenje, 31% razvija probleme s gutanjem i opstrukciju dišnih putova.
 Riža. trideset. Sonografska slika cističnog higroma vrata fetusa tijekom 16 tjedana trudnoće (u vratu fetusa se vidi velika tekuća tvorba - označena strelicom)
Riža. trideset. Sonografska slika cističnog higroma vrata fetusa tijekom 16 tjedana trudnoće (u vratu fetusa se vidi velika tekuća tvorba - označena strelicom)
načine. Pareza facijalnog živca zbog kirurškog liječenja javlja se u 24% bolesnika.
Opstetrička taktika je prekid trudnoće s ranom dijagnozom cistične higrome fetalnog vrata, s punom trudnoćom, porođaj se provodi kroz prirodni rodni kanal.
UROĐENE SRČANE GREŠKE
Učestalost prirođenih srčanih grešaka (CHD) kreće se od 1-2 do 8-9 na 1000 živorođene djece. Najčešći CHDs su atrijski i ventrikularni septalni defekti, otvoreni ductus arteriosus, stenoza plućne arterije, sindrom hipoplastičnog lijevog srca, jedna klijetka itd. U 90% slučajeva CHDs su posljedica multifaktorijalnog oštećenja (genetska predispozicija i okolišni čimbenici). Rizik od ponovne pojave mane je 2-5% nakon rođenja jednog i 10-15% nakon rođenja dvoje bolesne djece. Monogeno nasljeđivanje
starenje se opaža u 1-2% djece s prirođenom srčanom bolešću. Kromosomske abnormalnosti nalaze se u 5% djece, od kojih su glavne trisomije. U 1-2% novorođenčadi postoji kombinirani učinak različitih teratogena. Fetalna ehokardiografija je najinformativnija metoda za prenatalnu dijagnostiku kongenitalnih srčanih mana. Indikacije za prenatalnu dijagnostiku određene su stanjem majke i fetusa.
1. Indikacije zbog stanja majke:
Prisutnost CHD u članovima obitelji;
Dijabetes;
Uzimanje trudnih lijekova tijekom organogeneze;
Alkoholizam;
Sistemski eritematozni lupus;
Fenilketonurija.
2. Indikacije zbog stanja fetusa:
polihidramnion;
Ne-imuni vodena bolest;
Poremećaji srčanog ritma;
Ekstrakardijalni nedostaci;
Kromosomski poremećaji;
Simetrični oblik intrauterinog zastoja u rastu fetusa. Prognoza ovisi o vrsti defekta, prisutnosti popratnih anomalija i kromosomskih abnormalnosti.
Opstetrička taktika je da se nakon temeljite ehokardiografske studije izvodi kordo ili amniocenteza kako bi se dobio materijal za kromosomsku analizu. Ako se CHD otkrije u fetusu koji nije održiv, indiciran je prekid trudnoće. S punom terminskom trudnoćom, bolje je provesti porod u specijaliziranim perinatalnim centrima. Uz kombinirane nedostatke i genetske abnormalnosti, prekid trudnoće je neophodan u bilo kojem trenutku.
jednu komoru srca. Ovo je teška kongenitalna malformacija u kojoj su komore srca predstavljene jednom komorom ili velikom dominantnom komorom u kombinaciji sa zajedničkim atrioventrikularnim spojem koji sadrži dva atrioventrikularna ventila. Učestalost pojave defekta nije točno utvrđena. Jedna klijetka se lako dijagnosticira korištenjem standardnog četverokomornog dijela srca fetusa. Jedini
komora morfološki može biti desna i lijeva. Ukupna stopa preživljenja za sve tipove jedne klijetke u bolesnika bez kirurškog liječenja je 30%. Jedna klijetka često se kombinira s kromosomskim abnormalnostima, genskim poremećajima (Holt-Oramov sindrom), sindromom asplenije / polisplenije, koji se često formira u nekim bolestima majke, a također i na pozadini teratogenih učinaka retinoične kiseline. Prenatalni pregled kod pronalaska jedne klijetke treba uključiti kariotipizaciju i detaljan ultrazvučni pregled anatomije fetusa. Klinički tijek bolesti i taktika liječenja u neonatalnom razdoblju određeni su stanjem plućnog i sustavnog krvotoka.
Atrijski septalni defekt(AMPP) (slika 31). Predstavlja nedostatak septuma koji odvaja atrije. Uočava se u 17% svih prirođenih srčanih grešaka i njegova je najčešća strukturna anomalija. Često se kombinira s drugim intrakardijalnim anomalijama, kao i s neimunom fetalnom kapi. Možda kombinacija s kromosomskim abnormalnostima. Većina malih ASD-a ne otkriva se tijekom prenatalnog fetalnog ultrazvuka. Dijagnoza se može postaviti samo korištenjem višestrukih rezova i dopplerom u boji. Prenatalni pregled nakon otkrivanja ASD-a trebao bi uključivati
 Riža. 31. Sonografska slika opsežnog defekta atrijalnog septuma (označeno strelicom)
Riža. 31. Sonografska slika opsežnog defekta atrijalnog septuma (označeno strelicom)
započeti s određivanjem kariotipa i detaljnim proučavanjem ultrazvučne anatomije fetusa. Identifikacija izoliranog ASD-a u prenatalnom razdoblju ne zahtijeva promjenu taktike trudnoće i porođaja. U kasni datumi trudnoće potrebno je provesti dinamičku procjenu stanja fetusa.
Defekt ventrikularnog septuma(VMZHP). Predstavlja nedostatak septuma koji odvaja ventrikule. Prema lokalizaciji razlikuju se defekti gornjeg septuma (na razini mitralnog i trikuspidalnog zaliska), mišićnog dijela i izlaznog dijela septuma (subaortalni, subpulmonalni). Po veličini, VSD su podijeljeni u male (do 4 mm) i velike. VSD može biti izoliran ili kombiniran s drugim anomalijama, kromosomskim greškama i nasljednim sindromima. U općoj strukturi prirođenih srčanih grešaka, oko 20% je izolirana VSD, koja je najčešće dijagnosticirana greška. Učestalost malih, hemodinamski beznačajnih, mišićnih defekata doseže 53:1000 živorođene djece. Oko 90% takvih mana spontano se zatvori do 10. mjeseca života i ne utječe na prognozu života i zdravlja.
Većina malih VSD-ova nije otkrivena tijekom prenatalnog fetalnog ultrazvuka. Dijagnoza se može postaviti samo korištenjem višestrukih rezova i dopplerom u boji. Najčešće je VSD izoliran, ali se može kombinirati s kromosomskim abnormalnostima, genskim poremećajima, sindromima višestrukih malformacija. Prenatalni pregled za VSD trebao bi uključivati kariotipizaciju i detaljan pregled fetalne ultrazvučne anatomije. Identifikacija izoliranog VSD-a u prenatalnom razdoblju ne zahtijeva promjenu taktike trudnoće i porođaja. U kasnoj trudnoći potrebno je provesti dinamičku procjenu fetusa. Ako postoji sumnja na VSD, roditeljima treba pružiti sve informacije o prognozi za život i zdravlje nerođenog djeteta i obavijestiti pedijatra kako bi se osigurao odgovarajući nadzor novorođenčeta. Čak i s velikim VSD-om, bolest ponekad može biti asimptomatska i do 2-8 tjedana. U 50% slučajeva mali defekti se spontano zatvore prije navršenih 5 godina, a preostalih 80% nestanu u mladost. Većina bolesnika s nekompliciranim VSD ima dobru prognozu za život i zdravlje. Uz povoljan protok
bolesti značajna ograničenja tjelesne aktivnosti nije potrebna.
Ebsteinova anomalija- kongenitalna srčana bolest, karakterizirana abnormalnim razvojem i položajem listića trikuspidalnog ventila. S Ebsteinovom anomalijom, septalna i stražnja jedra trikuspidalnog ventila razvijaju se izravno iz endokarda desne klijetke srca, što dovodi do pomicanja abnormalnog ventila duboko u desnu klijetku i podjele klijetke na dva dijela: distalni (subvalvularni) - aktivni i proksimalni (supravalvularni ili atrijalizirani) - pasivni. Supravalvularni odjel, povezujući se s desnim atrijem, čini jednu funkcionalnu formaciju. Ebsteinova anomalija čini 0,5% svih urođenih srčanih mana. Ebsteinova anomalija može se lako dijagnosticirati pregledom standardnog fetalnog srca s četiri komore, jer je gotovo uvijek praćena kardiomegalijom. Prenatalna dijagnoza defekta temelji se na otkrivanju značajno povećanog desnog srca zbog desnog atrija. Ključna točka u dijagnozi Ebsteinove anomalije je vizualizacija pomaknutog trikuspidalnog zaliska na pozadini proširenog desnog atrija i normalnog miokarda desne klijetke. Važna prognostička vrijednost kod Ebsteinove anomalije je otkrivanje trikuspidalne regurgitacije tijekom Doppler ehokardiografije fetusa. Najraniji prenatalni ultrazvučna dijagnostika Ebsteinova anomalija je izvedena u 18-19 tjednu trudnoće. Prognoza za život s Ebsteinovom anomalijom obično je povoljna u slučajevima kada djeca prežive tijekom prve godine života bez kirurškog liječenja. Ebsteinova anomalija nije često u kombinaciji s kromosomskim aberacijama i sindromima višestrukih kongenitalnih malformacija. Ekstrakardijalne anomalije opažene su u 25%. Ishod u neonatalnom razdoblju ovisi o težini promjene na trikuspidalnom zalisku. Djeca s teškom insuficijencijom trikuspidalne valvule imaju visok postotak smrti. Klinički se insuficijencija trikuspidalne valvule očituje pojačanom cijanozom, acidozom i znakovima zatajenja srca. Kirurško liječenje je indicirano u bolesnika s izraženim simptomima bolesti koji ometaju normalan život djeteta. Operacija uključuje zatvaranje septuma
defekt, plastičnost trikuspidalnog zaliska i njegovo pomicanje na tipično mjesto. Bolnička smrtnost je 6,3%.
Tetralogija Fallot- složeni defekt koji uključuje nekoliko anomalija strukture srca: defekt ventrikularnog septuma, dekstropoziciju aorte, opstrukciju izlazne plućne arterije i hipertrofiju desne klijetke. U općoj strukturi prirođenih srčanih grešaka u živorođene djece Fallotova tetralogija iznosi 4-11%. Vrlo je teško dijagnosticirati Fallotovu tetradu kada se proučava četverokomorno srce fetusa. Kada se presječe kroz izlaze glavnih arterija, može se vidjeti tipičan subaortalni VSD i dekstrapozicija aorte. Važan dodatni kriterij je proširenje i pomak korijena aorte. Tetralogija Fallot je defekt plavog tipa, tj. u novorođenčadi, izražena cijanoza se utvrđuje u dobi od 6 tjedana do 6 mjeseci. Fallotova tetralogija odnosi se na srčane mane koje je teško dijagnosticirati i koje često ostaju neotkrivene ultrazvučnim probirom u razdoblju do 22 tjedna trudnoće. Najčešće se ovaj nedostatak dijagnosticira u trećem tromjesečju trudnoće ili nakon rođenja. Tetralogija Fallot ne zahtijeva posebne taktike upravljanja. Ako se otkrije ova patologija, potrebno je sveobuhvatno ispitivanje i prenatalno savjetovanje. Gotovo 30% živorođene djece s Fallot-ovom tetradom ima kombinirane ekstrakardijalne anomalije. Trenutno je opisano više od 30 sindroma višestrukih malformacija, čija struktura uključuje Fallotovu tetralogiju. Prenatalni pregled za otkrivanje Fallotove tetrade trebao bi uključivati određivanje kariotipa i detaljnu ultrazvučnu studiju anatomije fetusa. Prognoza za život u tetralogiji Fallot uvelike ovisi o stupnju opstrukcije izlaznog trakta desne klijetke. Više od 90% pacijenata koji su bili podvrgnuti potpunoj korekciji Fallotove tetrade prežive do odrasle dobi. Dugoročno se 80% pacijenata osjeća zadovoljavajuće i ima normalne funkcionalne parametre.
Transpozicija velikih arterija- bolest srca kod koje aorta, ili njen veći dio, izlazi iz desne klijetke, a plućna arterija iz lijeve klijetke. Čini 5-7% svih urođenih srčanih mana. Obično se ne dijagnosticira u prenatalnom razdoblju tijekom probira jer je proučavanje srca fetusa ograničeno na ispitivanje
samo rez s četiri komore. Da bi se identificirao nedostatak, potrebna je vizualizacija glavnih žila uz proučavanje njihovog položaja u odnosu jedna na drugu. Normalno, glavne arterije se križaju, a tijekom transpozicije napuštaju klijetke paralelno: aorta - iz desne klijetke, plućna arterija - s lijeve strane. Transpozicija glavnih arterija s netaknutim interventrikularnim i interatrijskim septama nije kompatibilna sa životom. Oko 8% živorođene djece s transpozicijom glavnih arterija ima udružene ekstrakardijalne anomalije. Prenatalni pregled trebao bi uključivati određivanje kariotipa i detaljnu ultrazvučnu studiju anatomije fetusa. Većina novorođenčadi s transpozicijom glavnih arterija i intaktnim interventrikularnim septumom ima izraženu cijanozu od prvih dana života. Kiruršku korekciju potrebno je provesti čim se otkrije neadekvatno miješanje krvotoka. Stopa smrtnosti novorođenčadi s ovom vrstom kirurškog liječenja je manja od 5-10%.
BOLESTI PRSNOG KOŠA
kongenitalna dijafragmalna hernija- defekt koji je posljedica usporavanja procesa zatvaranja pleuroperitonealnog kanala. Kod ovog defekta obično postoji nedovoljan razvoj posterolateralnog dijela lijeve polovice dijafragme. Nedostatak razdvajanja između trbušne šupljine i prsnog koša dovodi do pomicanja želuca, slezene, crijeva, pa čak i jetre u prsnu šupljinu, što može biti popraćeno pomakom medijastinuma i uzrokovati kompresiju pluća. Kao rezultat toga, često se razvija bilateralna hipoplazija pluća različite težine. Nerazvijenost pluća dovodi do abnormalnog formiranja krvožilnog sustava i sekundarne plućne hipertenzije. Kongenitalna dijafragmalna hernija javlja se u otprilike 1 od 2400 novorođenčadi.
Postoje četiri glavne vrste defekta: posterolateralna (Bochdalekova kila), anterolateralna, sternalna i Morgagnijeva kila. Bilateralne dijafragmalne kile čine 1% svih vrsta defekata. Pokret srca u desnu stranu prsa u kombinaciji s eho-negativnom strukturom (želudac) u njegovoj lijevoj polovici najčešće se dijagnosticira lijevostrana dijafragmalna hernija.
Kod kila s desne strane srce je obično pomaknuto ulijevo. U prsima se također mogu vizualizirati crijeva i jetra. Uz ovaj nedostatak često se bilježi polihidramnion. Kombinirane anomalije opažene su u 23% fetusa. Među njima prevladavaju urođene srčane mane, koje čine 16%. Dijagnoza mane može se provesti već u 14. tjednu trudnoće. Smrtnost kod kongenitalne dijafragmalne kile korelira s vremenom otkrivanja defekta: samo 33% novorođenčadi s defektom preživi u slučajevima kada je dijagnoza postavljena prije 25. tjedna, a 67% ako je kila otkrivena kasnije. Defekti dijafragme obično imaju multifaktorijalno podrijetlo, međutim, 12% slučajeva su u kombinaciji s drugim malformacijama ili su dio kromosomskih i nekromosomskih sindroma. Prenatalni pregled treba nužno uključiti određivanje fetalnog kariotipa i detaljan ultrazvučni pregled. Ako se otkriju kombinirane anomalije, diferencijalna dijagnoza može se provesti samo tijekom konzultacija uz sudjelovanje genetičara, sindromologa i pedijatara. Roditeljima treba savjetovati da se posavjetuju s pedijatrijskim kirurgom kako bi razgovarali o značajkama taktike liječenja u neonatalnom razdoblju, prognozi za život i zdravlje. Tijek neonatalnog razdoblja ovisi o težini hipoplazije pluća i težini hipertenzije. Veličina kile i volumen funkcionalnog plućnog tkiva također imaju značajan utjecaj na ishod u neonatalnom razdoblju. Abnormalni razvoj pluća može se predvidjeti u prisutnosti polihidramnija, dilatacije želuca, kao i pomicanja jetre fetusa u prsnu šupljinu. Prema literaturi, preživjelo je samo 22% djece s prenatalno postavljenom dijagnozom. Čak i kod izolirane kongenitalne dijafragmalne kile, samo 40% preživi. Neonatalna smrt obično je posljedica plućne hipertenzije i/ili respiratornog zatajenja.
ANOMALIJE FORMIRANJA STIJENKA TRBUŠNE ŠUPLJINE I RAZVOJNOG RAZVOJA GASTROINTESTINALNOG TRAKTA
Omfalokela (pupčana kila)(Slika 32). Nastaje kao posljedica nevraćanja organa trbušne šupljine iz amnionske šupljine kroz pupčani prsten. Omfalokela može uključivati bilo koji
 Riža. 32. Sonografska slika omfalokele (vizualizira se hernijalna vreća koja sadrži crijevne petlje i jetru)
Riža. 32. Sonografska slika omfalokele (vizualizira se hernijalna vreća koja sadrži crijevne petlje i jetru)
visceralni organi. Veličina hernialne formacije određena je njezinim sadržajem.
Prekrivena je amnioperitonealnom membranom, duž čije bočne površine prolaze žile pupkovine. Učestalost omfalokele je 1 na 3000-6000 novorođenčadi. Postoje izolirani i kombinirani oblici omfalokele. Ova patologija u 35-58% prati trisomija, 47% - kongenitalne srčane mane, 40% - malformacije genitourinarnog sustava, 39% - defekti neuralne cijevi. U 20% slučajeva otkriva se intrauterini zastoj u rastu.
Prenatalna ultrazvučna dijagnoza temelji se na otkrivanju okrugle ili ovalne formacije ispunjene trbušnim organima i neposredno uz prednji trbušni zid. Najčešće, sastav hernialnog sadržaja uključuje crijevne petlje i jetru. Pupkovina je pričvršćena izravno na hernialnu vrećicu. U nekim slučajevima prenatalna dijagnoza može se postaviti na kraju prvog tromjesečja trudnoće, iako se u većini slučajeva omfalokela otkrije u drugom tromjesečju. Prognoza ovisi o popratnim anomalijama. Perinatalni gubici češće su povezani s CHD, kromosomskim
aberacije i nedonoščad. Najveći defekt uklanja se jednofaznom operacijom, kod velike se izvode višefazne operacije za zatvaranje rupe u prednjem trbušnom zidu silikonskom ili teflonskom membranom. Opstetrička taktika određena je razdobljem otkrivanja defekta, prisutnošću kombiniranih anomalija i kromosomskih poremećaja. Ako se nedostatak otkrije u ranoj fazi trudnoće, treba je prekinuti. U slučaju otkrivanja popratnih anomalija koje nisu kompatibilne sa životom, potrebno je prekinuti trudnoću u bilo kojem trenutku. Način poroda ovisi o vitalnosti fetusa, budući da tijekom poroda s velikom omfalokelom može doći do puknuća hernialne vrećice i infekcije unutarnjih organa fetusa.
Gastroshiza- defekt prednjeg trbušnog zida u području pupka s eventeracijom crijevnih petlji prekrivenih upalnim eksudatom. Defekt se obično nalazi desno od pupka, hernialni organi nemaju membranu. Učestalost gastroshize je 0,94:10 000 novorođenčadi. Učestalost anomalije u trudnica mlađih od 20 godina veća je i iznosi 7 na 10 000 novorođenčadi.
Od kraja 70-ih. 20. stoljeće u Europi i SAD-u nastavlja se trend povećanja učestalosti rađanja djece s gastroshizom. Dodijelite izolirane i kombinirane oblike. Izolirana gastroshiza je češća i čini prosječno 79%. Kombinirani oblik otkriva se u 10-30% slučajeva i najčešće je kombinacija gastroshize s atrezijom ili intestinalnom stenozom. Među ostalim anomalijama, kongenitalne malformacije srca i mokraćnog sustava, sindrom orezati trbuh, hidrocefalus, nizak i polihidramnion.
Anomalija se javlja sporadično, ali postoje opažanja obiteljske bolesti s autosomno dominantnim tipom nasljeđivanja.
Najranija prenatalna ultrazvučna dijagnoza transvaginalnom ehografijom učinjena je u 12. tjednu trudnoće. U većini slučajeva dijagnoza se postavlja u drugom tromjesečju trudnoće, budući da je u ranim fazama (10-13 tjedana) moguća lažno pozitivna dijagnoza zbog prisutnosti fiziološke crijevne kile u fetusu. Prenatalna ultrazvučna dijagnoza gastroshize obično se temelji na vizualizaciji petlji crijeva u amnionska tekućina u blizini prednjeg trbušnog zida fetusa. Ponekad, osim crijevnih petlji, izvan
drugi organi mogu biti smješteni u trbušnoj šupljini. Točnost ultrazvučne dijagnoze gastroshize u II i III trimestru trudnoće varira od 70 do 95% i ovisi o gestacijskoj dobi, položaju fetusa, veličini defekta i broju organa koji se nalaze izvan prednje trbušne stijenke.
Ukupna prognoza za novorođenčad s izoliranom gastroshizom je dobra, s više od 90% dojenčadi preživi. Kod produljenja trudnoće, taktika upravljanja u drugom tromjesečju nema posebnih značajki. Zbog niske učestalosti kombinacije izolirane gastroshize s kromosomskim abnormalnostima, može se suzdržati od prenatalne kariotipizacije. U trećem tromjesečju trudnoće potrebno je provesti dinamičku procjenu funkcionalnog stanja fetusa, budući da je učestalost distresa kod gastroshize prilično visoka, a intrauterini zastoj u rastu formira se u 23-50% slučajeva.
Ako se gastroshiza otkrije prije održivosti fetusa, potrebno je izvršiti pobačaj. U donošenoj trudnoći porođaj se izvodi u ustanovi u kojoj je moguće kirurško zbrinuti.
duodenalna atrezija je najčešći uzrok opstrukcije tankog crijeva. Učestalost anomalije je 1:10 000 živorođene djece. Etiologija je nepoznata. Moguća je pojava defekta pod utjecajem teratogenih čimbenika. Opisana su obiteljska opažanja piloroduodenalne atrezije s autosomno recesivnim nasljeđivanjem. U 30-52% bolesnika anomalija je izolirana, au 37% nalaze se nedostaci razvoja koštanog sustava: abnormalni broj rebara, sacrum agenia, konjsko stopalo, bilateralna vratna rebra, bilateralna odsutnost prvih prstiju itd. U 2% kombinirana kombinacija abnormalnosti gastrointestinalnog trakta: nepotpuna rotacija želuca, jednjaka, ilijačnog crijeva i anusa, jetre, transakcije. al od živih Yeni. U 8-20% bolesnika otkrivaju se kongenitalne srčane mane, u otprilike 1/3 slučajeva atrezija dvanaesnika kombinirana je s trisomijom za 21 par kromosoma. Glavni prenatalni ehografski nalazi kod atrezije dvanaesnika su polihidramnion i klasični znak "dvostruki mjehurić" u fetalnom abdomenu. Slika "dvostrukog mjehurića" pojavljuje se kao rezultat širenja dijela dvanaesnika i želuca. Suženje između ovih tvorevina tvori pilorični dio želuca
ka i od velike je važnosti za točnu prenatalnu dijagnostiku ove mane. U velikoj većini slučajeva atrezija dvanaesnika dijagnosticira se u II i III tromjesečju trudnoće. U ranijim terminima dijagnoza ovog defekta predstavlja značajne poteškoće. Najranija dijagnoza duodenalne atrezije postavljena je u 14. tjednu.
Za određivanje opstetričke taktike provodi se detaljna ultrazvučna procjena anatomije unutarnjih organa fetusa i njegova kariotipizacija. Prije početka razdoblja održivosti fetusa indiciran je prekid trudnoće. Ako se izolirana anomalija otkrije u trećem tromjesečju, moguće je produljenje trudnoće, nakon čega slijedi porod u regionalnom perinatalnom centru i kirurška korekcija malformacije.
Izolirani ascites. Ascites je nakupljanje tekućine u peritonealnoj šupljini. Učestalost nije točno utvrđena. Ultrazvučnim pregledom fetusa ascites se očituje prisutnošću ehonegativnog prostora debljine 5 mm ili više u trbušnoj šupljini. U prenatalnom razdoblju ascites može biti izoliran ili biti jedan od znakova vodene bolesti neimunog podrijetla. Uz ascites, vodenu bolest fetusa karakterizira prisutnost potkožnog edema, pleuralnog i perikardijalnog izljeva, kao i povećanje debljine posteljice više od 6 cm, polihidramnija i hidrokele.
Ascites se može kombinirati s različitim strukturnim anomalijama, pa je indicirano temeljito ispitivanje svih unutarnjih organa fetusa. Među uzrocima izoliranog ascitesa treba razlikovati mekonijski peritonitis i kongenitalni hepatitis.
Do sada u literaturi nije bilo publikacija o otkrivanju izoliranog ascitesa u prvom tromjesečju trudnoće. Većina opažanja rane dijagnoze ascitesa javlja se na početku drugog tromjesečja trudnoće. Jedan od najčešćih uzroka neimune vodene bolesti su kromosomske abnormalnosti. S izoliranim ascitesom, kromosomske greške su rjeđe, ali se moraju uzeti u obzir kao moguća pozadina za razvoj ove patologije. Kada se otkrije ascites u fetusu, prvo je potrebno isključiti kombinirane anomalije i intrauterine infekcije. Tijek fetalnog ascitesa ovisi o njegovoj etiologiji. Idiopatski izolirani ascites ima povoljnu prognozu. U više od 50% promatranja zabilježen je njegov spontani nestanak. Najčešći uzrok izoliranog ascitesa je intrauterina infekcija.
parvovirus B19. Kod produljenja trudnoće potrebno je provesti dinamičko ehografsko promatranje, uključujući dopplersku procjenu protoka krvi u venskom kanalu. S normalnim vrijednostima protoka krvi u venskom kanalu u fetusa s ascitesom, u većini slučajeva primjećuje se povoljan perinatalni ishod. Kod povećanja ascitesa neki autori preporučuju terapijsku punkciju, osobito u slučajevima kada proces napreduje u kasnoj trudnoći. Glavna svrha punkcije je spriječiti diskordinaciju radna aktivnost i respiratorni distres u neonatalnom razdoblju. Ako se izolirani ascites otkrije u prenatalnom razdoblju i isključi se komorbiditet koji je nespojiv sa životom, dijete treba pažljivo dinamičko praćenje i simptomatsku terapiju nakon poroda.
Malformacije BUBREGA I MOKRAĆNOG TRAKTA
Renalna ageneza- potpuni nedostatak oba bubrega. Pojava defekta je posljedica kršenja sekvencijalnog lanca procesa normalne embriogeneze od pronefrosa do metanefrosa. Učestalost je u prosjeku 1:4500 novorođenčadi. Primjećuje se da se kod dječaka nalazi dvostruko češće. Patognomonični trijas ehografskih znakova renalne agenezije u fetusa predstavljen je odsutnošću njihovog odjeka i mokraćnog mjehura, kao i teškim oligohidramnijem. Oligohidramnion se odnosi na kasne manifestacije i može se otkriti nakon 16-18 tjedna trudnoće. Obično je bilateralna agenezija bubrega popraćena simetričnim oblikom sindroma zastoja u rastu fetusa. Bubrežna agenezija najčešće je sporadična, ali se može kombinirati s različitim anomalijama unutarnjih organa. Izravne posljedice oligohidramnija su hipoplazija pluća, deformacije kostura i lica, sindrom zastoja u rastu fetusa. Renalna agenezija opisana je u više od 140 sindroma višestrukih kongenitalnih malformacija, kromosomskih abnormalnosti i teratogenih učinaka. Nakon postavljanja dijagnoze, kariotipizaciju treba učiniti prenatalno ili nakon rođenja kako bi se isključile kromosomske abnormalnosti. U svim opažanjima bubrežne ageneze potreban je kompletan patoanatomski pregled. Prikazuje se ultrazvuk
pregledi bubrega u najbližoj rodbini. Uz prenatalno otkrivanje mane, treba preporučiti prekid trudnoće u bilo kojem trenutku. Ako obitelj odluči produljiti trudnoću, indicirana je konzervativna opstetrička taktika.
Autosomno recesivna policistična bolest bubrega (infantilni oblik). Očituje se obostranim simetričnim povećanjem bubrega kao rezultatom zamjene parenhima sekundarno proširenim sabirnim kanalom bez proliferacije vezivnog tkiva. Kreće se od klasične letalne varijante do infantilnih, juvenilnih, pa čak i odraslih oblika. U infantilnom obliku dolazi do sekundarne dilatacije i hiperplazije normalno formiranih sabirnih kanalića bubrega. Bubrezi su zahvaćeni simetrično, a cistične tvorevine su veličine 1-2 mm. Učestalost je 1,3-5,9:1000 novorođenčadi. Glavni ehografski kriteriji za malformacije su povećani hiperehogeni bubrezi, odsutnost eho mjehura i oligohidramnij. Povećanje veličine bubrega ponekad je toliko značajno da zauzimaju veliki dio poprečnog presjeka abdomena fetusa. Tipična ehografska slika može se pojaviti tek u trećem tromjesečju trudnoće. Prognoza je nepovoljna. Smrt dolazi od zatajenja bubrega. Opstetrička taktika je prekid trudnoće u bilo kojem trenutku.
Policistična bolest bubrega kod odraslih(autosomno dominantna bolest, hepatorenalna policistična bolest adultnog tipa, Potterov sindrom tipa III) karakterizira zamjena bubrežnog parenhima brojnim cistama različite veličine, koje nastaju zbog širenja sabirnih kanalića i drugih tubularnih segmenata nefrona. Bubrezi su zahvaćeni s obje strane i povećani, ali jednostrani proces može biti prva manifestacija bolesti. Jetra je također uključena u patološki proces - razvija se periportalna fibroza, koja ima žarišni karakter. Etiologija bolesti je nepoznata, ali tip nasljeđa uzrokuje 50% rizik od razvoja bolesti, a njezino genetsko žarište nalazi se na 16. paru kromosoma. Mutirani gen nosi jedna od 1000 osoba. Prodiranje gena događa se u 100% slučajeva, međutim, tijek bolesti može varirati od teških oblika sa smrtnim ishodom u neonatalnom razdoblju do asimptomatskih oblika koji se otkrivaju tek na autopsiji.
Policistična bolest bubrega(multicistična bolest, cistična bolest bubrega, Potterov tip II sindrom, displastična bolest bubrega) karakterizirana je cističnom degeneracijom bubrežnog parenhima zbog primarnog širenja bubrežnih tubula. Kod multicistične bubrežne displazije ureter i zdjelica su najčešće atrezirani ili ih nema. Proces može biti bilateralan, jednostran i segmentalan. Kod multicistične displazije bubreg je obično značajno povećan; uobičajeni oblik i normalno tkivo su odsutni. Bubreg je predstavljen višestrukim cistama s anehogenim sadržajem (slika 33).
 Riža. 33. Ehogram bilateralnih policističnih fetalnih bubrega (jako uvećani bubrezi koji sadrže više cista različitih promjera - označeno strelicom)
Riža. 33. Ehogram bilateralnih policističnih fetalnih bubrega (jako uvećani bubrezi koji sadrže više cista različitih promjera - označeno strelicom)
Veličine cista variraju u prilično širokom rasponu i ovise o trajanju trudnoće. Bliže terminu, promjer cista može doseći 3,5-4 cm.Mjehur se obično vizualizira s jednostranim procesom i ne vizualizira se s bilateralnim procesom. S bilateralnim procesom obično se bilježi oligohidramnion. Bolest se javlja uglavnom sporadično i može biti sekundarna u kombinaciji s drugim sindromima. akušerski
taktika u bilateralnom procesu dijagnosticiranom u ranim fazama, zbog nepovoljne prognoze, je prekid trudnoće. Uz jednostrani proces i normalan kariotip bez pridruženih anomalija, indiciran je konvencionalni porod, nakon čega slijedi konzultacija djeteta sa specijalistom.
Dilatacija urinarnog trakta. Anomalije genitourinarnog sustava u fetusu, popraćene ekspanzijom urinarnog trakta, mogu biti uzrokovane razni razlozi, uključujući vezikoureteralni refluks, idiopatsku pijelektazu, opstruktivne poremećaje, itd. S kliničkog gledišta, preporučljivo je razlikovati pijelektazu i opstruktivnu uropatiju u prenatalnom razdoblju.
Pijelektazija. Pijelektazija je karakterizirana prekomjernim nakupljanjem tekućine i širenjem fetalne bubrežne zdjelice.
Pijelektazija je najčešći nalaz fetalnog ultrazvuka. Učestalost njegovog razvoja nije utvrđena, budući da je ova patologija sporadična pojava. Nakon rođenja, dječacima se dijagnosticira 5 puta češće. U 27% djece s hidronefrozom otkriva se vezikoureteralni refluks, bilateralno udvostručenje uretera, bilateralni opstruktivni megaureter, nefunkcionalni kontralateralni bubreg i njegova ageneza, u 19% - anomalije u razvoju različitih organa. Za prenatalnu ultrazvučnu dijagnozu pijelektazije, bubrege fetusa treba pregledati i poprečno i uzdužno. O dilataciji bubrežne zdjelice prosuđuje se na temelju njezine anteriorno-posteriorne veličine pri poprečnom pregledu bubrega. Većina istraživača smatra pyelectasis proširenje bubrežne zdjelice u II tromjesečju trudnoće više od 5 mm, au III tromjesečju - više od 8 mm. S ekspanzijom bubrežne zdjelice fetusa preko 10 mm, uobičajeno je govoriti o hidronefrozi. Najčešća klasifikacija hidronefroze u fetusa je:
Stupanj I (fiziološka dilatacija):
Bubrežna zdjelica: anteriorno-posteriorna dimenzija<1 см;
Kortikalni sloj: nepromijenjen.
II stupanj:
Bubrežna zdjelica: 1,0-1,5 cm;
Šalice: nisu vizualizirane;
Kortikalni sloj: nepromijenjen.
III stupanj:
Bubrežna zdjelica: anteroposteriorna dimenzija >1,5 cm;
Čaške: blago proširene;
Kortikalni sloj: nepromijenjen.
IV stupanj:
Bubrežna zdjelica: anteroposteriorna dimenzija >1,5 cm;
Čaška: umjereno raširena;
Kortikalni sloj: malo promijenjen.
V. stupanj:
Bubrežna zdjelica: anteroposteriorna dimenzija >1,5 cm;
Čaške: jako proširene;
Kortikalni sloj: atrofija.
Proširenje bubrežne zdjelice fetusa može se promatrati s različitim kromosomskim abnormalnostima. Učestalost kromosomskih grešaka u fetusa s pijeloektazijom iznosi u prosjeku 8%. Kod većine fetusa s kromosomskim abnormalnostima otkriva se kombinacija pijelektazije i drugih razvojnih anomalija. Umjereno izražena pijelektazija ima dobru prognozu i rijetko se javlja potreba za kirurškim liječenjem nakon poroda. U većini opažanja primjećuje se spontano rješavanje umjereno teške pijelektazije nakon rođenja djeteta.
Opstetrička taktika ovisi o vremenu nastanka i trajanju tijeka patološkog procesa, kao io stupnju poremećene funkcije bubrega. Rani porod je opravdan s oligohidramnionom. U postnatalnom razdoblju prikazano je dinamičko praćenje i konzultacija s pedijatrijskim urologom.
opstruktivna uropatija. Opstrukcija urinarnog trakta u fetusa može se uočiti na bilo kojoj razini: visoka opstrukcija, opstrukcija na razini ureteropelvic fistule (PUR), opstrukcija na srednjoj razini (ureter), opstrukcija na razini vezikoureteralnog spoja (VUR), niska opstrukcija (uretra). URMS je najčešći uzrok opstruktivne uropatije u fetusa i čini prosječno 50% svih kongenitalnih uroloških anomalija. Glavne sonografske značajke OLMS uključuju dilataciju bubrežne zdjelice sa ili bez ekspanzije čašica; ureteri se ne vizualiziraju; mjehur može biti normalne veličine ili se u nekim slučajevima ne može vidjeti. Taktika u OLMS-u trebala bi biti očekivana. Ugradnja veziko-amnionskog šanta nije indicirana. Na ultrazvučni plač-
OPMS u fetusa uključuju dilataciju uretera i pijelektazu. Mokraćni mjehur je obično normalne veličine. Taktika vođenja je slična kao na OLMS-u. Najčešći uzrok niske opstrukcije su stražnji uretralni zalisci. S teškom opstrukcijom opaža se oligohidramnij koji dovodi do hipoplazije pluća, deformiteta struktura lica i udova, fibroze i displazije bubrežnog parenhima. Ehografsku sliku karakterizira prisutnost proširene uretre proksimalno od mjesta opstrukcije, izražena ekspanzija mjehura. Prenatalno liječenje niske opstrukcije ovisi o trajanju trudnoće, prisutnosti oligohidramnija i povezanih anomalija te funkcionalnom stanju bubrega. S umjereno teškom i neprogresivnom pijelektazom treba se pridržavati konzervativne taktike. S progresijom opstruktivnih poremećaja opravdan je porođaj s mogućom kirurškom korekcijom defekta kako bi se spriječili teški bubrežni poremećaji u fetusa. U prijevremenoj trudnoći u fetusa s teškom opstruktivnom uropatijom može se provesti intrauterina kirurška korekcija defekta.
KOŠTANI DEFEKT
Među prirođenim malformacijama koštanog sustava najčešća je amelija (aplazija svih udova); fokomelija (nerazvijenost proksimalnih udova, dok su ruke i stopala izravno povezani s tijelom); aplazija jedne od kostiju potkoljenice ili podlaktice; polidaktilija (povećanje broja prstiju na udovima); sindaktilija (smanjenje broja prstiju zbog fuzije mekih tkiva ili koštanog tkiva susjednih prstiju); abnormalna postavka zaustavljanja; osteohondrodisplazija, karakterizirana anomalijama u rastu i razvoju hrskavice i/ili kostiju (ahondrogeneza, ahondroplazija, tanatoformna displazija, osteogenesis imperfecta, hipofosfatazija itd.).
Najvažnija je dijagnoza nedostataka nespojivih sa životom. Mnogi oblici skeletne displazije kombiniraju se s hipoplazijom pluća zbog male veličine prsnog koša zbog nerazvijenosti rebara. Razvoj plućne insuficijencije u ovom slučaju može biti uzrok smrti djece u prvim satima izvanmaterničkog života.
Ahondroplazija je jedna od najčešćih nesmrtonosnih skeletnih displazija i uzrokovana je novom mutacijom u 90% slučajeva. Ahondroplazija je osteohondroplazija s defektima dugih kostiju i/ili aksijalnog skeleta. Učestalost je 0,24-5:10 000 poroda. Omjer muških i ženskih fetusa je 1:1. Skraćivanje kostiju kod ahondroplazije ne mora se pojaviti u fetusu do 24. tjedna trudnoće. Klasični ehografski nalazi uključuju kratke udove (manje od 5. percentila), mala prsa, makrocefaliju i sedlasti nos. Očekivano trajanje života s ahondroplazijom ovisi prvenstveno o vremenu kada mala veličina prsnog koša ne uzrokuje ozbiljne respiratorne probleme. Intelektualni razvoj kod defekta je normalan, ali postoji visok rizik od neuroloških poremećaja, posebice kompresije leđne moždine u razini foramena magnuma, što može ograničiti psihička vježba. Makrocefalija može biti posljedica blagog hidrocefalusa zbog male veličine foramena magnuma. Ahondroplazija je dobro proučen i čest tip kongenitalnog patuljastog rasta u novorođenčadi. Ozbiljne probleme mogu imati centralna i opstruktivna apneja za vrijeme spavanja. U dobi od 6-7 godina života često se bilježe kronične rekurentne infekcije srednjeg uha. U rano djetinjstvočesto postoji i zakrivljenost donjih ekstremiteta, što u teškim stanjima zahtijeva kiruršku korekciju. Obično visina odraslih osoba s ahondroplazijom varira od 106 do 142 cm.
ZDRAVO! U POZICIJI SAM 32-33 TJEDNA. KOD ZADNJEG ULTRAZVUČNOG NALAZA SŽS-A FETUSA: HIDROCEFALIJA, AGENEZIJA KALOZUMA, ARHODNA CISTA. MALI VSD U FETUSU. TIJEKOM TRUDNOĆE SAM BILA U BOLNICI 2 PUTA: U TERMINU OD 6-7 TJEDANA I 28-29 TJEDANA SA PRIJETNJAMA PRIJEVREMENOG POROĐAJA. TIJEKOM TRUDNOĆE URADJENA SU 3 SCREININGA, GDJE SE PLOD NORMALNO RAZVIJA BEZ POKAZATELJA CNS CM I DR. TEK SAM U 31-32 TJEDNU OTKRIO TAKVU DIJAGNOZU, KOJU SU POTVRDILA 3 AMERIČKA SPECIJALISTA. U ŠUPLJINI MATERNICE 1 FETUS U GLAVNOM PREDLEŽANJU. FETOMETRIJA: BRG-79MM(32TJEDNA) OG-285MM(31N.2DAN) OJ 257MM(31N.) DULJINA DESNOG I LIJEVOG HUMERUSA 54MM-31TJEDAN. 6D. DULJINA DESNE I LIJEVE FEMURALNE 56MM.-29TJEDAN 5 DANA DL. ITD. I LEV. KOSTI GLJEVNICE 51MM.-31TJEDAN DL. RADIUM BONE 44MM.- 30 TJEDANA 6 DANA PROCIJENJENA TEŽINA PLODA 1520 -+ 210GR. ANATOMIJA FETUSA: LATERALNE MOŽDANE KLIJETKE SU PROŠIRENE, ASIMETRIČNE, ROGOVI LATERALNIH KLIJETKI LIJEVO 17MM, DESNO 26MM. TREĆI VENTRIKUL JE DILATIRAN NA 10 MM. ŠUPLJINA TRANSPARENTNE PREGRADE SE NE VIZUALIZIRA. U SAGITALNOM PRESJEKU KALOZUM NIJE ZNAČAJNO LOCIRAN. SA STRANE OD SREDNJE CRTE, LIJEVO, UNUTAR PREDNJEG I SREDNJEG KRANIJALNOG STAVA, NALAZI SE ANEHOGENA STRUKTURA OBLIKA OKRUGLA VELIČINA 40*28 MM, AVASKULARNA U CDI MODUSU. VELIKI SPREMNIK 6 MM. MEĐUHEMISFERNA VELIČINA CERENERLUMA 36 MM – 30 TJEDNA.3 DANA. SYLVIANIN FUROUS JE LOCIRAN. STRUKTURA LICA: PROFIL-BEZ ZNAČAJKI, NOSOLABIČNI TROKUT-BEZ ZNAČAJKI. ŠIRINA ALVEOLARNOG NASTAVKA 27 MM. PROMJER OKA 15 MM, INTRAORBITALNA DIMENZIJA 17,5 MM. KRALJEŽNICA BEZ DEFORMACIJA. PLUĆA BEZ OBILJEŽJA. JEKE FETUSA. VRH SRCA SE NALAZI U PREDNJEM LIJEVOM KVADRANTU PRSNE ŠUPLJINE, BAZA RVF JE LIJEVO OD STERNUMA. VELIČINE SRCA PRAVILNE. Otkucaji srca 138 otkucaja. U MIN. OS SRCA POD KUTOM OD 45 STUPNJEVA U ODNOSU NA SAGITALNU RAVNINU. VELIČINE ATRIJUMA SU ISTE, ZALISAK OVALNOG OBLIKA OTVORENOG U ŠUPLJINU LIJEVOG ATRIJUMA, FUNKCIONIRA. VELIČINE KLIJETKE SU ISTE. U SREDNJOJ TREĆINI MR-a EHO-NEGATIVNA ZONA JE 1MM. ATRIVENTRIKULARNI ZALISCI SE NALAZE NA ISTOJ RAZINI I RADE SINKRONO. GRAĐA MIOKARDA, ENDOKARDIJA I PERIKARDIJA JE NORMALNA. ODSJECI KRATKE OSI KROZ GLAVNE ARTERIJE I PRESJEK KROZ 3 ŽILA BEZ ZNAČAJKI. U NAČINU CFM U ASCENDENTNOJ AORTI I PLUĆNOJ ARTERIJI TEČE LAMINAT. ŽELUDAC, ŽUČNI MJEHUR, CRIJEVA, BUBREZI, JETRA- BEZ OSOBINA. LOCIRA SE MJESTO PRIPAJANJA PUPČINE ZA PREDNJI TRBUŠNI STIJEND. PLACENTA SE NALAZI NA PREDNJIM STIJENKAMA MATERNICE, SEŽE DO DNA. DEBLJINA PLACENTE NORMALNA-33MM. STUPANJ zrelosti je 1. ŠTO ODGOVARA DATUMU TRUDNOĆE. KOLIČINA OKOLNE VODE JE NORMALNA. PUPČINA IMA 3 ŽILE, PRIČVRŠĆENE PARACENTRALNO. STIJENKE MATERNICE BEZ OBILJEŽJA. POREMEĆAJ UTERO-PLACENTARNOG I FETUSNO-PLACENTARNOG KRVOTOKA SE NE UOČAVA. ŠTO OVO DIJETE IMA, ŠTO DA OČEKUJEM I ŠTO UČINITI?
Urođene malformacije fetusa uključuju sljedeće anomalije koje se javljaju kod nerođenog djeteta:
Odsutnost mozga (anencefalija);
- otvoreni oblik kile leđne moždine (leđna bifida);
- kongenitalne malformacije mokraćnog sustava fetusa;
- bolesti srca u fetusu ili patološke promjene u kardiovaskularnom sustavu;
- razne anomalije u razvoju udova u fetusa - atrezija (odsutnost udova);
- rascjep usne i nepca, drugi maksilofacijalni deformiteti.
Zašto dolazi do kongenitalnih malformacija fetusa
Pojava i razvoj raznih mana ploda može nastati kao posljedica utjecaja velikog broja čimbenika od kojih je većina do sada ostala nerazjašnjena.
Prema etiološkim znakovima, sve kongenitalne malformacije fetusa dijele se na:
Odstupanja u kromosomskim setovima roditelja (nasljedna);
- embrij ili fetus je oštećen izloženošću pesticidima, lijekovima ili infekcijama (teratogeni);
- zajednički utjecaj na nerođeno dijete genetskih i okolišnih čimbenika, koji pojedinačno ne mogu biti uzrok defekta (multifaktorijalni).
Prema nekim podacima, onečišćenje biosfere također može biti uzrok bolesti u 70% slučajeva, razvoja patologija u 60%, a smrti djece u 50% slučajeva.
Urođene malformacije fetusa i kasniji abnormalni razvoj djece nakon rođenja također su povezani s profesionalna djelatnost- ako osoba dulje vrijeme doživljava emocionalni stres, izloženost prašini, visokoj ili niske temperature, stalno u kontaktu s proizvodima kemijska industrija ili soli teških metala.
Također ako buduća mama pati od značajne pretilosti, to može biti ozbiljan uzrok abnormalnosti u razvoju neuralne cijevi fetusa. Takve promjene u malom tijelu fetusa mogu biti uzrokovane ne samo prekomjernom težinom trudnice, već i njezinim oštrim smanjenjem rani datumi trudnoća.
fetalne malformacije i naknadnu trudnoću
Mnogi fetalni CM se mogu liječiti. Nakon rođenja, ovisno o postojećoj anomaliji, dijete prolazi niz potrebnih postupaka za njezinu korekciju ili liječenje i nastavlja normalan život. U slučaju kada je CM fetusa nespojiv sa životom djeteta izvan maternice, trudnoća se prekida. Šest mjeseci nakon ovog zahvata možete planirati sljedeću trudnoću. Ponekad se paru savjetuje da pričeka godinu dana. Tijekom tog vremena budući roditelji podvrgavaju se određenim genetskim testovima i studijama, prema rezultatima kojih će liječnik odrediti kada je moguće začeti dijete.
U pripremi za sljedeću trudnoću, par mora izbjegavati utjecaj negativnih čimbenika, Zdrav stil životaživota, uzmite multivitamine da ojačate svoje tijelo.
16769 0
Kongenitalne malformacije fetusa zauzimaju 2.-3. mjesto u strukturi uzroka perinatalne smrtnosti, a po učestalosti posljednjih godina značajno porasla. U tom smislu, rana dijagnoza malformacija je od posebne važnosti, pridonoseći pravodobnom rješavanju pitanja mogućnosti daljnjeg produljenja trudnoće, što je, pak, određeno vrstom anomalije, kompatibilnošću sa životom i prognozom postnatalnog razvoja.
Klasifikacija uobičajenih malformacija središnjeg živčani sustav(CNS) može se predstaviti na sljedeći način:
1. Hidrocefalus:
a) stenoza cerebralnog akvadukta;
b) otvoreni hidrocefalus;
c) Dandy-Walkerov sindrom.
2. Papiloma koroidnog pleksusa.
3. Defekti neuralne cijevi:
a) spina bifida;
b) anencefalija;
c) cefalokela.
4. Mikrocefalija.
Hidrocefalus javlja se s učestalošću od 0,3-0,8 na 1000 živorođene djece. U većini slučajeva, kongenitalni hidrocefalus uzrokovan je opstrukcijom jednog od odjeljaka puta cirkulacije cerebrospinalne tekućine (likvora). Često se hidrocefalus kombinira s drugim anomalijama: u 37% slučajeva prati ga druga intrakranijalna patologija - hipoplazija corpus callosum, cefalocela, arteriovenske anomalije, arahnoidne ciste; ekstrakranijalne anomalije - u 63%. Od potonjih valja istaknuti malformacije bubrega (jednostrana i bilateralna agenezija i displazija), srčane mane (ventrikularni septalni defekt, Fallot tetralogija), meningomijelokelu, rascjep gornje usne, tvrdog i mekog nepca, agenezu anusa i debelog crijeva, disgenezu gonada, Meckeleov sindrom. Kromosomske abnormalnosti nalaze se u 11% fetusa - trisomija 21 para, uravnotežene translokacije, mozaicizam.
Hidrocefalus je predstavljen u tri glavna oblika:
- stenoza cerebralnog akvadukta;
- otvoreni hidrocefalus;
- Dandy-Walkerov sindrom.
Stenoza akvadukta mozga(SVM) je oblik opstruktivnog hidrocefalusa uzrokovan suženjem Silvijevog akvadukta. Specifična učestalost SVM-a doseže 43%, omjer muške i ženske djece je 1:8. Anomalija ima polietiološki karakter: genetski, infektivni, teratogeni i tumorski čimbenici, među kojima prevladavaju infektivni (50%). U eksperimentalnim studijama potvrđena je uloga toksoplazmoze, sifilisa, citomegalovirusne infekcije, zaušnjaka i gripe.
U određenom dijelu opažanja uzrok stenoze cerebralnog akvedukta je genetska patologija, koji se može naslijediti u recesivnom tipu vezan za X kromosom. Spolno povezano nasljeđe smatra se rijetkim uzrokom WMS-a, budući da se javlja s učestalošću od 1 slučaja na 200 braće i sestara probanda s hidrocefalusom. Međutim, moguće je da je ovaj tip nasljeđa 25% među muškom djecom. Pretpostavlja se da gliom, meningeom, neurofibromatoza i tuberozna skleroza dovode do stenoze akvadukta zbog mehanizma kompresije, a otvoreni hidrocefalus - kao rezultat edema bijele tvari i vanjske kompresije. Kombinirane anomalije javljaju se u 16% djece.
Dijagnoza ovog oblika hidrocefalusa temelji se na detekciji ultrazvukom ekspanzije lateralnih i III ventrikula s nepromijenjenim dimenzijama IV ventrikula. Potrebno je provesti temeljito snimanje fetalne kralježnice kako bi se isključile anomalije povezane sa spolom (slika 1).
Riža. 1. Trudnoća 21 tjedan. Opstruktivni hidrocefalus
Prognoza: smrtnost u dječjoj dobi kreće se od 11-30%; intelektualni razvoj može biti normalno.
Opstetrička taktika: prije nego što fetus postigne sposobnost preživljavanja, indiciran je prekid trudnoće; kod postavljanja dijagnoze u kasnijim fazama, način poroda određuje se isključivo opstetričkim indikacijama.
Otvoreni hidrocefalus(OG) - proširenje ventrikula mozga i njegovog subarahnoidnog sustava kao posljedica opstrukcije ekstraventrikularnog sustava izlaznih puteva cerebrospinalne tekućine.
Otvoreni hidrocefalus drugi je najčešći i čini 38% svih slučajeva hidrocefalusa. Etiologija OH nije razjašnjena. OH se otkriva u djece s defektima kralježnice i obliteracijom prednjeg sagitalnog sinusa, subarahnoidalnim krvarenjima, papilomom horoidnog pleksusa i odsutnošću Paccionijevih granulacija. Subarahnoidalno krvarenje je najčešći uzrok otvorenog hidrocefalusa u novorođenčadi; u prenatalnom razdoblju izuzetno je rijetka. Također se rijetko nasljeđuje, iako učestalost pojavljivanja doseže 1-2%, što je znatno više nego u općoj populaciji.
Patogeneza: mehanička opstrukcija ekstraventrikularnog sustava mozga i poremećena reapsorpcija likvora dovode do širenja subarahnoidalnog prostora, a potom i moždanih komora; unutarnji hidrocefalus razvija se u pozadini začepljenja cerebralnog akvadukta, zbog povećanog intrakranijskog tlaka.
Prenatalna dijagnoza OH provodi se dinamičkim ultrazvučnim skeniranjem. U ovom slučaju, patognomonični znak je proširenje subarahnoidne cisterne.
Prognoza: stopa smrtnosti doseže 11%. Većina preživjele djece zadržala je normalnu inteligenciju. Kada se OH kombinira s defektima neuralne cijevi ili papilomom horoidnog pleksusa, prognoza je nepovoljnija.
Opstetričke taktike: u ranim fazama otkrivanja OH, indiciran je prekid trudnoće, s punom trudnoćom, porođaj se provodi kroz prirodni rodni kanal.
Za dandy-walker sindrom tipična je kombinacija sljedećih anomalija:
1) hidrocefalus različitih stupnjeva;
2) ciste stražnje lubanjske jame;
3) defekti cerebelarnog vermisa, kroz koje cista komunicira sa šupljinom IV ventrikula.
Sindrom se javlja u 12% djece s kongenitalnim hidrocefalusom. Etiologija je nepoznata. Ovaj sindrom može biti jedna od manifestacija genetskih bolesti (Meckel i Warburg sindrom), a također se može otkriti u kromosomskim aberacijama (Turnerov sindrom, 6p-, 9gh+, trisomija 9, triploidija). U rijetkim slučajevima moguće je autosomno recesivno nasljeđivanje s rizikom recidiva do 25%.
Patoembriogeneza. Prema teoriji Dandy-Walker, atrezija Luschkinih i Magendiejevih otvora obično dovodi do širenja ventrikularnog sustava. Sindrom je složena anomalija u razvoju središnjih struktura mozga u području romboidne jame. Hipoplazija cerebelarnog vermisa i ciste stražnje jame javljaju se sekundarno, zbog kompresije njegovom oštro proširenom IV klijetkom. Postoji i neravnoteža u proizvodnji cerebrospinalne tekućine u lateralnim, III i IV ventrikulima. Defekt moždanog crva varira od potpune aplazije do blagog cijepanja. Unatoč činjenici da je hidrocefalus glavni dijagnostički znak Dandy-Walkerovog sindroma, većina djece ga nema u trenutku rođenja, ali se manifestira u prvim mjesecima djetetovog života.
Sindrom je često (u više od 50% slučajeva) u kombinaciji s drugim malformacijama središnjeg živčanog sustava (agenezija corpus callosum, encefalokela), malformacijama bubrega (policistični) i srca (ventrikularni septalni defekt).
Dijagnostika: ova anomalija je naznačena otkrivanjem cističnih formacija u stražnjoj lubanjskoj jami tijekom ehografije; patognomoničan akustični znak sindroma je defekt u moždanom crvu kroz koji cista komunicira s četvrtom klijetkom.
Prognoza nepovoljna: smrtnost doseže 50%, 50-60% preživjele djece ima zaostajanje u intelektualnom razvoju.
Opstetričke taktike: abortus u bilo kojem trenutku.
Papiloma koroidnog pleksusa(PSS). Intrakranijska neoplazma pojavljuje se s učestalošću od 0,6% svih tumora mozga otkrivenih u odraslih i 3% u djece. Papiloma se može lokalizirati u bilo kojem dijelu ventrikularnog sustava, ali se češće nalazi na razini vestibula lateralnih ventrikula. Karakteristična je jednostrana lokalizacija tumora, iako nije isključen bilateralni proces. Najčešće je PSS predstavljen vilusnim tkivom, histološki sličnim tkivu intaktnog horoidnog pleksusa i benigno je. Međutim, moguća je malignost tumora s klijanjem u susjedno živčano tkivo. Papiloma koroidnog pleksusa obično se kombinira s hidrocefalusom.
Etiologija nepoznato. U svjetskoj praksi poznata su izolirana opažanja PSS-a u bolesnika s Aicardijevim sindromom (ova je bolest povezana s kromosomom X i karakterizirana je agenezom žuljevitog tijela, korioretinalnim prazninama, anomalijama kralježnice, epilepsijom i mentalnom retardacijom).
PSS se dijagnosticira u djece s hidrocefalusom tijekom neurosonografije ili rendgenskih zraka. U djetinjstvu se metodom izbora treba smatrati kontrastom kompjutorizirana tomografija. Ultrazvučno skeniranje je najinformativnija metoda prenatalne dijagnoze. Ehografski kriteriji PSS-a su asimetrija oblika i veličine kontralateralnih ventrikula, vizualizacija u predvorju lateralnih ventrikula slabo ehogenih formacija uz normalni koroidni pleksus. Prenatalno, PSS III i IV ventrikula nije otkriven.
Metoda izbora za PSS terapiju je kirurško uklanjanje tumori. S benignom prirodom procesa, kirurško liječenje može imati povoljan ishod, ali operacija je tehnički složena i popraćena je velikim gubitkom krvi. S malignom lezijom (u više od 20% slučajeva), prognoza je nepovoljna. Smrtnost u PSS-u doseže 35%, a 72% preživjele djece ima različite stupnjeve težine nedostataka u mentalnom i mentalnom razvoju.
Opstetričke taktike: porod vodi kroz prirodni rodni kanal; primjena vaginalnih operacija je kontraindicirana. Porod se preporučuje u velikim perinatalnim centrima, gdje se može pružiti hitna neonatološka i pedijatrijska neurokirurška skrb. Pitanje korištenja carskog reza za smanjenje rizika od porođajne traume s intrakranijalnim krvarenjem nije konačno riješeno.
Odabrana predavanja iz opstetricije i ginekologije
ur. A.N. Strizhakova, A.I. Davidova, L.D. Belotserkovceva
Relevantnost. Prenatalna dijagnostika (PD) kongenitalnih malformacija fetusa iznimno je važna komponenta antenatalne skrbi koja omogućuje prevenciju rađanja djece s teškim, nekorektivim malformacijama, sa socijalno značajnim fatalnim genskim i kromosomskim bolestima. To je glavni čimbenik u smanjenju morbiditeta, invaliditeta i mortaliteta. Osim toga, prenatalno otkrivanje kongenitalnih anomalija mozga u fetusa utječe na opstetričku taktiku i smanjuje negativne psihološke i socijalne posljedice za majku i obitelj u cjelini, budući da su kongenitalne malformacije (KM) mozga općenito teške i neizlječive bolesti (rano i kvalitetno dijagnosticiranje malformacija CNS-a može objektivno predvidjeti situaciju i pridonijeti pravi izbor taktika liječenja). Dakle, kompetentna prenatalna dijagnostika, u kombinaciji s prikupljanjem cjelovitih informacija usmjerenih na prepoznavanje različitih čimbenika rizika, omogućuje liječniku i rizičnoj trudnici adekvatan pristup pitanju poroda.
Metode, koji se koristi za antenatalnu (prenatalnu) dijagnozu, preporučljivo je podijeliti na neizravne, kada je predmet studije trudnica, i izravne, kada se ispituje sam fetus. Potonji mogu biti invazivni i neinvazivni.
Neizravne metode uključuju proučavanje opstetričke i ginekološke povijesti, medicinsko genetsko savjetovanje, kao i bakteriološke i serološke studije. Zasebno treba reći o provođenju biokemijskih testova probira usmjerenih na procjenu razine fetoproteina, estriola, humanog korionskog gonadotropina itd. Treba napomenuti da je glavna svrha neizravnih metoda odabir žena iz visokorizičnih skupina za daljnje dubinsko praćenje već na razini centara za reproduktivno zdravlje. Glavne indikacije za upućivanje trudnice na PD približno su iste u cijelom svijetu. To uključuje:
[1
] dob žene iznad 35 godina;
[2
] prisutnost najmanje dva spontani pobačaji(abortusi) u ranoj trudnoći;
[3
] prisutnost u obitelji djeteta ili fetusa iz prethodne trudnoće s Downovom bolešću, drugim kromosomskim bolestima, s višestrukim kongenitalnim malformacijama, obiteljskim nositeljstvom kromosomskih preraspodjela;
[4
] mnoge monogene bolesti prethodno dijagnosticirane u obitelji ili bliskim rođacima;
[5
] primjena niza farmakoloških pripravaka prije i u ranoj fazi trudnoće;
[6
] prošle virusne infekcije (hepatitis, rubeola, toksoplazmoza itd.);
[7
] zračenje jednog od supružnika prije začeća.
pročitajte i članak: Toksoplazmoza(na web stranicu)
Glavne neinvazivne metode su [ 1 ] ultrazvučni (sonografski) pregled (ultrazvuk) i [ 2 ] magnetska rezonancija (MRI).
Ultrazvučni pregled središnjeg živčanog sustava i leđne moždine u fetusa jedan je od najvažnijih i najodgovornijih zadataka prenatalne ehografije, budući da značajno utječe na optimizaciju opstetričke taktike i odluku roditelja o produljenju ili prekidu trudnoće. Optimalno vrijeme ehografskog pregleda fetusa za isključivanje većine defekata i bolesti središnjeg živčanog sustava je:
[1
] 11 - 14 tjedana trudnoće;
[2
] 19 - 21 tjedan trudnoće;
[3
] 30 - 33 tjedna trudnoće.
To odgovara početnim fazama manifestacije različitih skupina defekata i bolesti središnjeg živčanog sustava, a također osigurava kontinuitet dijagnoze i općeprihvaćenih standarda vođenja trudnoće i poroda. U isto vrijeme, shema ultrazvučnog pregleda fetusa trebala bi uključivati procjenu ehografske anatomije fetusa s proučavanjem kostiju svoda lubanje, glavnih struktura mozga, okovratnog prostora, profila, orbite, nosnih kostiju, orijentacije srca, kralježnice, prednjeg trbušnog zida, želuca, crijeva, mjehura i ekstremiteta.
Podaci brojnih autora svjedoče o visokoj dijagnostičkoj vrijednosti ultrazvuka u prenatalnoj dijagnostici kongenitalnih malformacija, što omogućuje identificiranje više od 80-90% malformacija fetusa tijekom trudnoće. Značajan dio grube patologije razvoja fetusa može se otkriti u prvom tromjesečju, ovisno o vremenu pregleda i svim zahtjevima protokola probira za procjenu anatomije fetusa.
Korištenje suvremenih ultrazvučnih strojeva ekspertne klase, transvaginalna ehografija u 3/4D modovima značajno povećava točnost dijagnoze. Proučavanje fetalnog mozga u srednjoj sagitalnoj ravnini, što je postalo praktično moguće u većini slučajeva pomoću trodimenzionalne ehografije, omogućuje diferencirani pristup procjeni norme i patologije središnjih moždanih struktura. Aksijalni presjeci fetalnog mozga korišteni u rutinskoj ultrazvučnoj praksi nisu omogućili dobivanje jasne slike corpus callosuma. Proširenjem protokola prve studije probira s procjenom "intrakranijalne prozirnosti" povećana je točnost dijagnosticiranja spine bifide u prvom tromjesečju trudnoće.
Korištenje color Doppler mapiranja za vizualizaciju cerebralnih žila u fetusu u ranoj ontogenezi pouzdano omogućuje vizualizaciju žila koje opskrbljuju određenu strukturu 2-5 tjedana ranije od njihove standardne ehografske detekcije već na kraju 1. i na početku 2. tromjesečja trudnoće. Sveobuhvatna procjena razvoja brazda i subarahnoidnih prostora omogućuje dijagnosticiranje kršenja formiranja cerebralnog korteksa već u drugom tromjesečju trudnoće. Anomalije u razvoju središnjeg živčanog sustava, kao što su kršenja embriogeneze i rane fetalne organogeneze, mogu se otkriti pomoću moderne ehografije do 21 tjedna trudnoće. Istodobno, ehografski znakovi destruktivnih lezija i volumetrijskih formacija CNS-a mogu se dijagnosticirati samo u II i III tromjesečju trudnoće (S.M. Voevodin, 2012).
Transvaginalna ehografija s proučavanjem anatomskih struktura fetusa ultrazvukom u 11-14 tjednu trudnoće vrlo je informativna metoda prenatalne dijagnoze u ranoj trudnoći, koja omogućuje otkrivanje više od polovice svih kongenitalnih malformacija, koje su u većini slučajeva grube.
Također je potrebno napomenuti vrijednost za dijagnozu kongenitalnih malformacija takvog ultrazvučnog znaka kao polihidramnija. Ozbiljnost polihidramnija korelira s učestalošću kongenitalnih malformacija fetusa. Uspostavljen je izravan odnos između amnionska tekućina te učestalost kongenitalnih malformacija fetusa.
Općenito, uspjeh dijagnoze također ovisi o vrsti kongenitalnih malformacija (brojne anomalije je prilično teško dijagnosticirati), gestacijskoj dobi u kojoj se provodi studija, količini amnionske tekućine i konstitucionalnim karakteristikama pacijenta (teška pretilost stvara poteškoće u transabdominalnom skeniranju). Ostali čimbenici koji otežavaju ispravno tumačenje rezultata probira mogu biti gestacijska dob, višestruka trudnoća, etnička pripadnost roditelja, dijabetes kod majke. Prenatalna dijagnoza ageneze corpus callosum (čiji su glavni uvjeti otkrivanja II-III tromjesečja trudnoće), kila kralježnice prilično je komplicirana. U isto vrijeme, unatoč visokoj točnosti i relativnoj jednostavnosti ultrazvučne dijagnostike takvih apsolutno smrtonosnih defekata kao što su akranija i anencefalija, neki od njih se otkrivaju nakon 24 tjedna trudnoće, što može ukazivati na nedovoljno visoku kvalifikaciju liječnika, nepoštivanje uvjeta pregleda i metoda pregleda, kasno upućivanje žena na drugu ili treću razinu pregleda. Još jedan negativan čimbenik može biti odbijanje žena od prenatalne kariotipizacije. Osim toga, takva pitanja prenatalne ultrazvučne dijagnoze kongenitalnih malformacija središnjeg živčanog sustava kao diferencijalna dijagnoza s rijetkim nedostacima.
MRI je visoko informativna metoda prenatalne dijagnoze i može se koristiti kod sumnje na fetalne malformacije tijekom ultrazvučnog pregleda, posebno u slučajevima anomalija CNS-a. U drugom i trećem tromjesečju trudnoće, korištenje MRI kao dodatnog pregleda poboljšava dijagnozu defekata CNS-a i omogućuje razjašnjenje dijagnoze u slučaju patologije cerebralnog korteksa, srednjih struktura mozga, stražnje lubanjske jame i poremećaja dinamike CSF-a. MRI se može uspješno koristiti u slučajevima kada rezultati ultrazvuka nisu dovoljno informativni. Međutim, iako MR može potvrditi UZ dijagnozu i pružiti detaljnije podatke, relativno visoka cijena, nedostatak standardiziranih referentnih vrijednosti i ograničena dostupnost MR razlozi su zašto UZ ostaje studija izbora za dijagnosticiranje fetalne CM.
pročitajte takođerčlanak "MRI mozga fetusa: pregled" S. Yazbek i P.E. Grant. Neurographics, svezak 5, broj 5, 1. rujna 2015., str. 181-191(11) [1. dio] i [2. dio]
Oko Je li magnetska rezonanca sigurna tijekom trudnoće? možeš čitati
Proučavanje markerskih embrionalnih proteina u krvnom serumu majke. Posljednjih godina posebno važnu ulogu ima proučavanje markerskih embrionalnih proteina u krvnom serumu majke, kao što su alfa-fetoprotein (AFP), korionski gonadotropin (hCG), slobodni estradiol i neki drugi. Svrha takvih studija je identificirati žene s visokim rizikom da imaju djecu s urođenim i nasljednim manama. Promjene u serumskim markerima tipične su za 90,9% žena s kongenitalnim malformacijama središnjeg živčanog sustava. Provedeno u optimalnom vremenu (15-16 tjedana trudnoće) pomoću tri testna sustava, istraživanje omogućuje prepoznavanje do 80% fetusa s nedostacima u razvoju unutarnjih organa i do 65% s kromosomskim bolestima.
Prenatalna laboratorijska dijagnostika anomalija neuralne cijevi temelji se prvenstveno na određivanju razine fetalnog AFP-a. Ovaj protein je glavna komponenta proteinskog sustava fetalnog seruma i određuje se 30. dana gestacije. Povećanje razine AFP-a u amnionskoj tekućini znak je otvorenog defekta neuralne cijevi. U drugom tromjesečju trudnoće ultrazvuk može pouzdano otkriti abnormalnost mozga fetusa. Budući da u stvaranju estriola sudjeluju i fetus i posteljica, razina estriola može poslužiti kao idealan pokazatelj funkcioniranja fetoplacentarnog sustava. Što je niža razina hormona, to je veća vjerojatnost razvoja patološkog stanja fetusa.
Istodobno, tumačenje izoliranih rezultata biokemijskog testiranja može biti teško. Uzimajući u obzir krivulje distribucije vrijednosti glavnih markera, postoji velika zona preklapanja između norme i patologije, što ne dopušta korištenje samo jednog markera, potreban je njihov puni kompleks: hCG, AFP i estriol.
Biokemijsko testiranje u prvom tromjesečju trudnoće, koje uključuje [ 1 ] određivanje koncentracije progesterona, [ 2 ] nekonjugirani estriol, [ 3 ] β-frakcija humanog korionskog gonadotropina (β-hCG) i [ 4 ] proteina povezanih s trudnoćom (7 - 8 ili 11 - 12 tjedana) je više učinkovita metoda prenatalni probir nego tradicionalni "trostruki" test drugog tromjesečja, tj. AFP, β-hCG, estriol - u 16 - 17 tjednu trudnoće.
Unatoč prilično visokoj učinkovitosti suvremenih neinvazivnih metoda, dovoljno potpune informacije o kariotipu embrija, biokemijskim i genotipskim značajkama njegovih stanica mogu se dobiti samo na temelju odgovarajućih studija tkiva samog fetusa ili njegovih privremenih organa (placente, koriona), dobivenih invazivnim putem u bilo kojoj fazi trudnoće. Amniocenteza se najčešće provodi za otkrivanje kromosomskih abnormalnosti i genetskih mutacija, ali amnionska tekućina također se može koristiti za dijagnosticiranje defekata neuralne cijevi. Najčešći moguće rizike postupci su spontani pobačaj (0,5% do 1,0%), mrlje nakon zahvata, infekcija, pucanje ovoja i ozljeda ili gubitak fetusa.
Najinformativniji je sveobuhvatan pregled trudnica primjenom suvremenih ultrazvučnih tehnologija u kombinaciji s biokemijskim probirom, što povećava točnost dijagnosticiranja kongenitalnih malformacija središnjeg živčanog sustava fetusa u ranim fazama drugog tromjesečja. Osim toga, identifikacija znakova fetalne hipoksije može ukazivati na moguću prisutnost ne samo fetoplacentalne insuficijencije, već i kongenitalne patologije, budući da hipoksija i prenatalni razvoj značajno pogoršati prognozu za kongenitalne malformacije kompatibilne sa životom.
Treba zapamtiti, Što kongenitalne anomalije mozga otkrivene prenatalno ili u neonatalnom razdoblju, ne računajući nepostojane vanjske malformacije, [ !!! ] NE MOŽE imati nikakve specifične kliničke simptome:
[1 ] u neurosonografskim i magnetskim rezonantnim studijama, osim specifičnih nozoloških oblika kongenitalnih malformacija mozga (Chiarijev sindrom, Dandy-Walkerov sindrom, okluzivni hidrocefalus i dr.), treba obratiti pozornost na hipoplazije, koje su mnogo češće;
[2 ] u nedostatku prenatalne i neonatalne dijagnostike pomoću ultrazvuka i magnetske rezonancije nastaju uvjeti za kasnu dijagnozu, u vrijeme kada neuropsihijatrijski simptomi dolaze na prvo mjesto u kliničkoj slici, a prisutnost mentalne retardacije, intrakranijalne hipertenzije može poslužiti kao osnova za postavljanje dijagnoza kao što su cerebralna paraliza, hidrocefalus i dr.
pročitajte i članak: Neurosonografija: Ultrazvučna dijagnostika perinatalnih lezija CNS-a(na laesus-de-liro.live-journal.com)
pročitajte i članak: Dandy Walkerov sindrom(na web stranicu)
pročitajte i članak: Anomalija (malformacija) Chiari tip I(na web stranicu)
pročitajte i članak: Cerebralna paraliza(na web stranicu)
Najveća informativna vrijednost konvulzivnog sindroma ukazuje na prisutnost agenezije corpus callosuma ili agirije, hipertenzivno-hidrocefalnog sindroma na prisutnost kongenitalnog hidrocefalusa, a sindroma motoričkih poremećaja na prisutnost spinalne hernije.
Zaključak. Kako bi se spriječilo rađanje djece s defektima središnjeg živčanog sustava (neuralne cijevi), trenutno se koriste sljedeće metode: [ 1 ] primarni test probira za otkrivanje strukturnih abnormalnosti fetusa, uključujući otvoreni/zatvoreni defekt neuralne cijevi (anencefalija, encefalokela, spina bifida) - ultrazvuk fetusa u drugom tromjesečju trudnoće; [ 2 ] određivanje razine majčinog serumskog AFP-a; [ 3 ] genetsko savjetovanje slučajeva s pozitivnim rezultatima probira u vrijeme defekata neuralne cijevi (ultrazvuk + majčin serum AFP); [ 4 ] antenatalna MRI kao dodatna metoda snimanja fetusa; [ 5 ] dijagnostička amniocenteza za procjenu kariotipa fetusa, određivanje razine AFP u amnionskoj tekućini i aktivnosti acetilkolinesteraze; [ 6 ] pri potvrđivanju prisutnosti defekata neuralne cijevi u fetusa, obitelji treba ponuditi mogućnosti vođenja prave trudnoće - kako produljenje s mogućnošću pre- ili postnatalne korekcije defekta, tako i prekid trudnoće s defektom nespojivim sa životom; način poroda odabire se pojedinačno - to može biti i prirodni (vaginalni) porod (u nedostatku kontraindikacija za takav) s praćenjem otkucaja srca fetusa i porod carskim rezom; [ 7 ] postporođajne konzultacije za informiranje žene o uzroku nastanka defekta neuralne cijevi u fetusa, mogućnosti ponavljanja takvog stanja u fetusu tijekom sljedećih trudnoća i prevenciji, posebno preporuci uzimanja folne kiseline od 5000 mcg dnevno od strane roditelja 3 mjeseca prije začeća i tijekom prvog tromjesečja trudnoće.







