Les malformations congénitales du fœtus occupent la 2e à la 3e place dans la structure des causes de décès périnatal du fœtus et du nouveau-né. Le diagnostic précoce des malformations, qui est nécessaire pour résoudre rapidement le problème de la possibilité de prolonger la grossesse, qui est déterminé par le type de défaut, la compatibilité avec la vie et le pronostic du développement postnatal, est d'une grande importance. Selon l'étiologie, on distingue les malformations congénitales héréditaires (génétiques), exogènes et multifactorielles du fœtus. Héréditaire comprennent les malformations résultant de mutations, c'est-à-dire changements persistants dans les structures héréditaires des gamètes ou du zygote. Selon le niveau auquel la mutation s'est produite (gènes ou chromosomes), on distingue les syndromes monogéniques et les maladies chromosomiques. Les défauts exogènes comprennent les défauts causés par l'effet néfaste de facteurs exogènes. Ces facteurs, agissant pendant la période de gamétogenèse ou de grossesse, conduisent à la survenue de malformations congénitales sans perturber la structure de l'appareil héréditaire.
Les malformations d'origine multifactorielle sont appelées défauts apparus sous l'influence combinée de facteurs génétiques et exogènes. Il existe également des défauts isolés (localisés dans un organe), systémiques (au sein d'un système organique) et multiples (dans les organes de deux systèmes ou plus).
DÉFAUTS DU SYSTÈME NERVEUX CENTRAL
Classement des malformations les plus courantes du système nerveux central :
1. Hydrocéphalie :
Sténose de l'aqueduc cérébral;
Hydrocéphalie ouverte ;
Syndrome de Dandy-Walker.
2. Papillome du plexus choroïde.
3. Anomalies du tube neural :
- spina bifida;
anencéphalie ;
Céphalocèle.
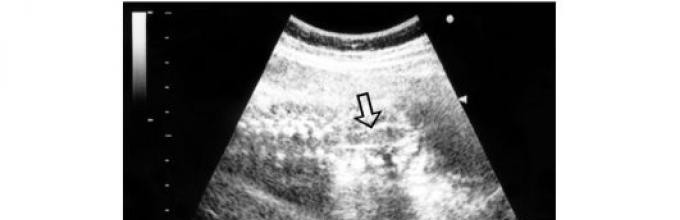
4. Microcéphalie. Hydrocéphalie
Hydrocéphalie- une augmentation de la taille des ventricules du cerveau avec une augmentation simultanée de la pression intracrânienne, accompagnée dans la plupart des cas d'une augmentation de la taille de la tête (Fig. 28).
Riz. 28. Image échographique d'hydrocéphalie fœtale sévère (les flèches indiquent des ventricules cérébraux fortement dilatés, dont le cortex est considérablement aminci, la taille de la tête fœtale dépasse les valeurs normales pour un âge gestationnel donné)
La ventriculomégalie est une augmentation isolée de la taille des ventricules, non accompagnée d'une augmentation de la taille de la tête. L'hydrocéphalie est observée avec une fréquence de 0,1 à 2,5 pour 1000 nouveau-nés. Environ 60 % des fœtus atteints d'hydrocéphalie sont des garçons. L'hydrocéphalie peut être le résultat de nombreuses maladies d'étiologies diverses. Dans la plupart des cas, il se développe à la suite d'une violation de l'écoulement du liquide céphalo-rachidien. La forme communicante de l'hydrocéphalie est causée par une
obstruction culaire, tandis que la forme obstructive est une obstruction intraventriculaire. Parfois, l'hydrocéphalie entraîne une augmentation de la production de liquide céphalo-rachidien (par exemple, dans le contexte du papillome des plexus vasculaires) ou une violation de sa réabsorption dans l'espace sous-arachnoïdien.
Des anomalies extracrâniennes dans l'hydrocéphalie surviennent dans 63 % des cas : agénésie et dysplasie des reins, communication interventriculaire, tétralogie de Fallot, méningomyélocèle, dédoublement la lèvre supérieure, palais mou et dur, atrésie de l'anus et du rectum, dysgénésie gonadique. L'hydrocéphalie est représentée principalement par une sténose de l'aqueduc cérébral (rétrécissement de l'aqueduc sylvien) ; hydrocéphalie ouverte (expansion des ventricules du cerveau et du système sous-arachnoïdien du cerveau à la suite de l'obstruction du système extraventriculaire des voies d'écoulement du liquide céphalo-rachidien); Syndrome de Dandy-Walker (association d'hydrocéphalie, de kystes de la fosse crânienne postérieure, de défauts du vermis cérébelleux, à travers lesquels le kyste communique avec la cavité du ventricule IV). Lorsqu'une hydrocéphalie est détectée, l'anatomie des structures cérébrales, ainsi que la colonne vertébrale, doivent être soigneusement évaluées pour exclure spina bifida. Un examen complet du fœtus devrait inclure un examen échocardiographique, car l'hydrocéphalie est souvent associée à des malformations cardiaques congénitales. Avec l'hydrocéphalie avant la période de viabilité fœtale, il est conseillé de discuter de la question de l'interruption de grossesse avec les parents. Lors de la prolongation de la grossesse, une surveillance échographique dynamique est affichée toutes les 2 semaines. Avec une augmentation de l'hydrocéphalie à l'atteinte de la maturité des poumons du fœtus, la question de l'accouchement précoce et du shunt doit être posée. L'efficacité du pontage ventriculaire prénatal n'a pas encore été prouvée et cette opération est peu utilisée. césarienne montré uniquement avec une macrocéphalie sévère et l'absence d'autres malformations. En présence d'anomalies macroscopiques combinées qui aggravent le pronostic vital, l'opération de choix est la céphalocentèse.
Anomalies du tube neural. Ce terme comprend l'anencéphalie, le céphalocèle et spina bifida.
spina bifida- une anomalie dans le développement de la colonne vertébrale, résultant d'une violation du processus de fermeture du tube neural (Fig. 29).
La sortie par un défaut des membranes de la moelle épinière est appelée méningocèle. Si le sac herniaire contient du tissu nerveux, la formation
 Riz. 29. Image échographique spina bifida dans la colonne lombo-sacrée (marquée d'une flèche)
Riz. 29. Image échographique spina bifida dans la colonne lombo-sacrée (marquée d'une flèche)
est appelée méningomyélocèle. Distinguer spina bifida kystique(forme kystique de hernie rachidienne avec formation d'un sac herniaire contenant les membranes du cerveau et/ou la substance du cerveau) et spina bifida occulte(forme cachée, qui ne s'accompagne pas de la formation d'une saillie herniaire). Le plus souvent, ce défaut est localisé dans la colonne lombaire et sacrée. Fréquence d'occurrence spina bifida dépend de la zone géographique. Dans certaines régions du Royaume-Uni, la fréquence de ce défaut est de 4 pour 1000 nouveau-nés. Aux États-Unis, ce chiffre est de 0,5 pour 1000, bien que cela dépende des caractéristiques raciales et géographiques. spina bifida- une malformation qui survient en raison d'une violation de la fermeture du tube neural à la 4ème semaine de développement embryonnaire. Cette anomalie est héritée de manière multifactorielle. spina bifide a peut se former à la suite d'une hyperthermie maternelle, si elle souffre de diabète sucré, d'une exposition à des facteurs tératogènes, et également faire partie de syndromes génétiques (avec un gène mutant isolé) ou d'anomalies chromosomiques (trisomie de 13 et 18 paires de chromosomes, triploïdie , translocation déséquilibrée ou chromosome en anneau). La hernie vertébrale est associée à plus
plus de 40 syndromes de malformations multiples (hydrocéphalie, malformations cardiaques congénitales et système génito-urinaire).
L'examen prénatal comprend la détermination du caryotype et une échographie approfondie. Attention particulière doit être donnée à l'anatomie de la tête, du cœur, des bras et des jambes. Si une méningomyélocèle est détectée avant la viabilité du fœtus, la femme doit se voir proposer une interruption médicale de grossesse. Lors de la prolongation de la grossesse, une échographie dynamique est indiquée toutes les 2 à 3 semaines pour évaluer l'apparition d'autres signes (par exemple, la ventriculomégalie). Les parents doivent recevoir une consultation avec un neurochirurgien pour discuter des possibilités d'intervention chirurgicale après l'accouchement (fermeture du défaut ou pontage), ainsi que du pronostic pour la vie et la santé de l'enfant. L'accouchement doit être effectué dans les grands centres périnataux dès que les poumons du fœtus atteignent une maturité suffisante. Risque empirique de récidive spina-bibida est de 3 à 5 %. L'utilisation de fortes doses d'acide folique (4 mg), commencée 3 mois avant la grossesse prévue et poursuivie pendant la première moitié de celle-ci, peut réduire considérablement le risque d'anomalie.
Toute anomalie ouverte du tube neural doit être fermée dans les 24 premières heures de vie. Un traitement antibactérien commencé immédiatement après la naissance peut réduire le risque de complications infectieuses. Le pronostic vital et sanitaire dépend du niveau de localisation de la méningomyélocèle, ainsi que du nombre et de la nature des anomalies associées. développement mental les enfants qui ont un tour de tête normal et un cerveau bien formé à la naissance ne souffrent pas. Les patients atteints de méningomyélocèle situés en L2 et au-dessus sont presque toujours contraints d'utiliser un fauteuil roulant.
Anencéphalie(pseudocéphalie, dysencéphalie extracrânienne) - l'absence des hémisphères cérébraux et de la majeure partie de la voûte crânienne, alors qu'il existe un défaut dans l'os frontal au-dessus de la région supraorbitaire, le temporal et une partie de l'os occipital sont absents. La partie supérieure de la tête est recouverte d'une membrane vasculaire. Les structures du mésencéphale et du diencéphale sont partiellement ou complètement détruites. L'hypophyse et la fosse rhomboïde sont majoritairement préservées. Les manifestations typiques comprennent des yeux exorbités, une grande langue et un cou court. Cette pathologie survient avec une fréquence de 1 sur 1000. Plus souvent elle
trouvé chez les filles nouveau-nées. Acranie(exencéphalie) - l'absence de la voûte crânienne en présence d'un fragment de tissu cérébral. C'est une pathologie plus rare que l'anencéphalie. L'anencéphalie est le résultat de l'échec de la fermeture du neuropore rostral dans les 28 jours suivant la fécondation. Hérédité multifactorielle et autosomique récessive, des anomalies chromosomiques sont notées. Les facteurs de risque comprennent le diabète maternel. Dans des expériences sur des animaux, la tératogénicité des radiations, des salicylates, des sulfamides et des niveaux élevés de dioxyde de carbone a été établie. Le diagnostic échographique peut être établi dès 12-13 semaines de grossesse. L'anencéphalie et l'acranie sont des malformations absolument mortelles, par conséquent, dans les deux cas, la femme doit se voir proposer une interruption de grossesse. Tous les nouveau-nés atteints d'anencéphalie et d'acranie meurent dans les 2 semaines suivant la naissance. Le risque empirique de récidive de l'anencéphalie est de 3 à 5 %. L'utilisation de fortes doses d'acide folique (4 mg), commencée 3 mois avant la grossesse prévue et poursuivie pendant la première moitié de celle-ci, peut réduire considérablement le risque d'anomalie.
Céphalocèle(encéphalocèle, méningocèle crânienne ou occipitale, division du crâne) - renflement du contenu du crâne par un défaut osseux. Le terme "méningocèle crânien" désigne la saillie uniquement à travers le défaut des membranes méningées. Lorsque le tissu cérébral se trouve dans le sac herniaire, le terme "encéphalocèle" est utilisé. La céphalocèle est rare (1:2000 naissances vivantes) et est une composante de nombreux syndromes génétiques (syndromes de Meckele, dédoublement médian du visage) et non génétiques (constriction amniotique). Le céphalocèle se développe à la suite de la non-fermeture d'une anomalie du tube neural et survient à la 4ème semaine de développement. Un défaut dans le crâne, à travers lequel les membranes du cerveau et du tissu cérébral peuvent prolapsus, se forme à la suite de la non-séparation de l'ectoderme de surface et du neuroectoderme sous-jacent. Si un céphalocèle est détecté, la femme doit se voir proposer une interruption médicale de grossesse. Lors de la prolongation de la grossesse, la tactique d'accouchement dépend de la taille et du contenu du sac herniaire. À grandes tailles défaut, prolapsus d'une quantité importante de tissu cérébral, ainsi qu'en présence de microcéphalie et d'hydrocéphalie, le pronostic pour la vie et la santé est extrêmement défavorable.
L'accouchement par césarienne n'est pas indiqué dans ces observations. Il est possible de recommander la décompression du sac herniaire pour créer les conditions d'un accouchement par le canal génital naturel. Une césarienne peut être recommandée s'il y a un petit défaut et si le sac herniaire est petit.
Microcéphalie (microencéphalie) est un syndrome clinique caractérisé par une diminution du périmètre crânien et un retard mental. Elle survient avec une fréquence de 1 nouveau-né sur 1360, avec des anomalies combinées de 1,6 pour 1000 naissances vivantes. La microcéphalie est une maladie polyétiologique, dans le développement de laquelle des facteurs génétiques (aberrations chromosomiques, défauts monogéniques) et environnementaux jouent un rôle important. Le pronostic dépend de la présence d'anomalies associées. La trisomie sur les chromosomes 13, 18, le syndrome de Meckel sont des lésions fatales. L'examen prénatal doit comprendre la détermination du caryotype fœtal et une échographie approfondie. En l'absence d'anomalies concomitantes, le pronostic dépend de la taille de la tête : plus elle est petite, plus l'indice de développement intellectuel est faible. La microcéphalie est une maladie incurable. Tactiques obstétriques - interruption de grossesse avant que le fœtus n'atteigne la viabilité.
ANOMALIES DES STRUCTURES FACIALES ET DU COU
Visage fendu(fente labiale et palatine) est un défaut linéaire s'étendant du bord de la lèvre à l'ouverture nasale.
La fente palatine, associée à la fente labiale, à travers les processus alvéolaires et le palais dur peut se propager à la cavité nasale ou même au bas de l'orbite. Une fente labiale bilatérale est observée chez 20%, une fente labio-palatine - 25%. Avec une lésion unilatérale, la fente est plus souvent située à gauche. La fente faciale représente environ 13% de toutes les malformations et est enregistrée avec une fréquence de 1:800 naissances vivantes. Les garçons ont plus souvent des fentes que les filles. Les anomalies combinées sont retrouvées dans 50 % des cas avec une fente palatine isolée et seulement dans 13 % avec une fente labio-palatine. Les structures faciales se forment entre la 4e et la 10e semaine de grossesse. Les structures frontonasales non appariées fusionnent avec les structures appariées maxillaire et mandibulaire.
mes tubercules. Dans ces observations, lorsque le processus de fusion n'est pas terminé, des fentes se forment. En règle générale, il est possible de diagnostiquer une fente faciale uniquement au deuxième trimestre de la grossesse avec une échographie de dépistage. La détection prénatale d'un défaut par échographie est difficile, cependant, grâce à l'échographie et à la cartographie Doppler couleur, les possibilités de son diagnostic s'élargissent. L'échographie Doppler peut visualiser le mouvement du liquide à travers le nez, la bouche et le pharynx. En présence d'une fente, la nature du mouvement du fluide change. L'échographie tridimensionnelle peut clarifier le diagnostic dans ces observations lorsqu'une fente a été suspectée dans une étude bidimensionnelle, mais sa visualisation claire n'a pas été obtenue. Il est possible de diagnostiquer une anomalie à l'aide de la fœtoscopie, y compris l'embryoscopie. En l'absence d'anomalies associées, des tactiques obstétriques généralement acceptées sont utilisées, quel que soit le moment du diagnostic. Prendre de l'acide folique avant et pendant la prochaine grossesse peut réduire le risque de fentes.
Le dédoublement de la lèvre supérieure (fente labiale) n'interfère pas avec l'acte de succion et n'est qu'un défaut esthétique. Avec une combinaison de fractionnement de la lèvre supérieure, de la mâchoire et du palais dur (fente palatine), des troubles fonctionnels sont notés: lors de la succion, le lait s'écoule par le nez en raison de sa communication avec la cavité buccale; le lait peut être inhalé. Le pronostic est favorable : les méthodes chirurgicales modernes permettent de réaliser la correction des défauts esthétiques et fonctionnels.
hygroma kystique(lymphangiome ou conséquences d'une obstruction du tronc lymphatique jugulaire) est une accumulation de liquide enkysté (Fig. 30). Elle se caractérise par la présence de kystes uniques ou multiples des tissus mous dans le cou, résultant de troubles du système lymphatique. Les hygromes kystiques surviennent avec une fréquence de 1:200 fausses couches spontanées (taille coccyx-pariétale du fœtus supérieure à 30 mm). L'hygroma kystique est souvent associé à des aberrations chromosomiques (syndrome de Turner, trisomie de 13, 18, 21 paires de chromosomes, mosaïcisme). En tant qu'anomalie isolée, elle est héritée de manière autosomique récessive. Pronostic : dans la plupart des cas, le fœtus meurt au cours des deux premiers trimestres de la grossesse. Environ 90% nécessitent un traitement chirurgical, 31% développent des problèmes de déglutition et une obstruction des voies respiratoires.
 Riz. trente. Image échographique d'hygroma kystique du cou fœtal au cours d'une grossesse de 16 semaines (une grande formation de liquide est visualisée dans le cou du fœtus - indiquée par une flèche)
Riz. trente. Image échographique d'hygroma kystique du cou fœtal au cours d'une grossesse de 16 semaines (une grande formation de liquide est visualisée dans le cou du fœtus - indiquée par une flèche)
façons. Une parésie du nerf facial due à un traitement chirurgical survient chez 24 % des patients.
La tactique obstétricale consiste à mettre fin à la grossesse avec un diagnostic précoce de l'hygroma kystique du cou du fœtus, avec une grossesse à terme, l'accouchement est effectué par le canal de naissance naturel.
MALADIES CARDIAQUES CONGÉNITALES
La fréquence des malformations cardiaques congénitales (CHD) varie de 1-2 à 8-9 pour 1000 naissances vivantes. Les coronaropathies les plus fréquentes sont les communications interauriculaires et ventriculaires, la persistance du canal artériel, la sténose de l'artère pulmonaire, l'hypoplasie du cœur gauche, le ventricule unique, etc. Dans 90 % des cas, les coronaropathies résultent d'une atteinte multifactorielle (prédisposition génétique et facteurs environnementaux). Le risque de récidive du défaut est de 2 à 5 % après la naissance d'un enfant et de 10 à 15 % après la naissance de deux enfants malades. Hérédité monogénique
le vieillissement est observé chez 1 à 2 % des enfants atteints de cardiopathie congénitale. Des anomalies chromosomiques sont retrouvées chez 5% des enfants, dont les trisomies sont les principales. Chez 1 à 2% des nouveau-nés, il existe un effet combiné de divers tératogènes. L'échocardiographie fœtale est la méthode la plus informative pour le diagnostic prénatal des cardiopathies congénitales. Les indications du diagnostic prénatal sont déterminées par l'état de la mère et du fœtus.
1. Indications dues à l'état de la mère :
La présence de coronaropathies chez les membres de la famille ;
Diabète;
Prendre des médicaments enceintes pendant l'organogenèse;
Alcoolisme;
Le lupus érythémateux disséminé;
Phénylcétonurie.
2. Indications dues à l'état du fœtus :
polyhydramnios;
hydropisie non immunitaire ;
Troubles du rythme cardiaque ;
Défauts extracardiaques ;
Troubles chromosomiques ;
Forme symétrique du retard de croissance intra-utérin du fœtus. Le pronostic dépend du type de défaut, de la présence d'anomalies concomitantes et d'anomalies chromosomiques.
La tactique obstétricale est qu'après une étude échocardiographique approfondie, une cordo ou une amniocentèse est effectuée afin d'obtenir du matériel pour l'analyse chromosomique. Si une maladie coronarienne est détectée chez un fœtus non viable, l'interruption de grossesse est indiquée. Avec une grossesse à terme, il est préférable d'effectuer l'accouchement dans des centres périnataux spécialisés. Avec des défauts combinés et des anomalies génétiques, l'interruption de grossesse est nécessaire à tout moment.
seul ventricule du cœur. Il s'agit d'une malformation congénitale sévère dans laquelle les ventricules du cœur sont représentés par une chambre unique ou un grand ventricule dominant en combinaison avec une jonction auriculo-ventriculaire commune contenant deux valves auriculo-ventriculaires. La fréquence d'apparition du défaut n'a pas été déterminée avec précision. Le ventricule unique est facilement diagnostiqué à l'aide d'une section standard à quatre chambres du cœur fœtal. Le seul
le ventricule morphologiquement peut être à la fois droit et gauche. Le taux de survie global pour tous les types de ventricule unique chez les patients sans traitement chirurgical est de 30 %. Le ventricule unique est souvent associé à des anomalies chromosomiques, des troubles génétiques (syndrome de Holt-Oram), un syndrome d'asplénie/polysplénie, souvent formé dans certaines maladies maternelles, et également dans le contexte des effets tératogènes de l'acide rétinoïque. L'examen prénatal lorsqu'un seul ventricule est trouvé doit inclure un caryotypage et un examen détaillé de l'anatomie échographique du fœtus. L'évolution clinique de la maladie et les tactiques de prise en charge en période néonatale sont déterminées par l'état du flux sanguin pulmonaire et systémique.
Communication interauriculaire(AMPP) (fig. 31). Représente une déficience du septum séparant les oreillettes. On l'observe dans 17 % de toutes les malformations cardiaques congénitales et c'est son anomalie structurelle la plus courante. Souvent associé à d'autres anomalies intracardiaques, ainsi qu'à une hydropisie fœtale non immunitaire. Peut-être une combinaison avec des anomalies chromosomiques. La plupart des petits TSA ne sont pas détectés lors de l'échographie fœtale prénatale. Le diagnostic ne peut être fait qu'à l'aide de plusieurs coupes et d'une imagerie Doppler couleur. L'examen prénatal lors de la détection d'un TSA devrait inclure
 Riz. 31. Image échographique d'une communication interauriculaire étendue (indiquée par une flèche)
Riz. 31. Image échographique d'une communication interauriculaire étendue (indiquée par une flèche)
pour commencer la détermination du caryotype et une étude détaillée de l'anatomie échographique du fœtus. L'identification d'un TSA isolé dans la période prénatale ne nécessite pas de changement de tactique de grossesse et d'accouchement. DANS dates tardives grossesse, une évaluation dynamique de l'état du fœtus doit être effectuée.
Communication interventriculaire(VMZHP). Représente une déficience du septum séparant les ventricules. Selon la localisation, on distingue les défauts du septum supérieur (au niveau des valves mitrale et tricuspide), de la partie musculaire et de la section de sortie du septum (sous-aortique, sous-pulmonaire). Par taille, les VSD sont divisés en petits (jusqu'à 4 mm) et grands. Le VSD peut être isolé ou associé à d'autres anomalies, anomalies chromosomiques et syndromes héréditaires. Dans la structure générale des malformations cardiaques congénitales, environ 20 % sont des VSD isolées, qui sont les malformations les plus fréquemment diagnostiquées. La fréquence des petits défauts musculaires hémodynamiquement insignifiants atteint 53:1000 naissances vivantes. Environ 90% de ces défauts se ferment spontanément à l'âge de 10 mois et n'affectent pas le pronostic pour la vie et la santé.
La plupart des petits VSD ne sont pas détectés lors de l'échographie fœtale prénatale. Le diagnostic ne peut être fait qu'à l'aide de plusieurs coupes et d'une imagerie Doppler couleur. Le plus souvent, la VSD est isolée, mais peut être associée à des anomalies chromosomiques, des troubles géniques, des syndromes de malformations multiples. L'examen prénatal pour un VSD doit inclure un caryotypage et un examen détaillé de l'anatomie fœtale par ultrasons. L'identification d'un VSD isolé dans la période prénatale ne nécessite pas de changement dans la prise en charge de la grossesse et de l'accouchement. En fin de grossesse, une évaluation dynamique du fœtus doit être réalisée. En cas de suspicion de VSD, les parents doivent recevoir des informations complètes sur le pronostic pour la vie et la santé de l'enfant à naître et en informer le pédiatre pour assurer une surveillance adéquate du nouveau-né. Même avec de gros VSD, la maladie peut parfois être asymptomatique pendant 2 à 8 semaines. Dans 50% des cas, les petits défauts se referment spontanément avant l'âge de 5 ans, et les 80% restants disparaissent en adolescence. La plupart des patients atteints de VSD non compliqué ont un bon pronostic pour la vie et la santé. Avec un débit favorable
maladie des restrictions importantes sur l'activité physique n'est pas nécessaire.
Anomalie d'Ebstein- cardiopathie congénitale, caractérisée par un développement et une localisation anormaux des feuillets de la valve tricuspide. Avec l'anomalie d'Ebstein, les voiles septales et postérieures de la valve tricuspide se développent directement à partir de l'endocarde du ventricule droit du cœur, ce qui entraîne un déplacement de la valve anormale profondément dans le ventricule droit et une division du ventricule en deux sections : distale (sous-valvulaire) - actif et proximal (supravalvulaire ou auriculaire) - passif. Le service supravalvulaire, relié à l'oreillette droite, forme une formation fonctionnelle unique. L'anomalie d'Ebstein représente 0,5 % de toutes les malformations cardiaques congénitales. L'anomalie d'Ebstein peut être facilement diagnostiquée en examinant un cœur fœtal standard à quatre cavités, car elle est presque toujours accompagnée d'une cardiomégalie. Le diagnostic prénatal de l'anomalie est basé sur la détection d'un cœur droit considérablement élargi en raison de l'oreillette droite. Le point clé dans le diagnostic de l'anomalie d'Ebstein est la visualisation d'une valve tricuspide déplacée sur fond d'oreillette droite dilatée et de myocarde ventriculaire droit normal. Une valeur pronostique importante dans l'anomalie d'Ebstein est la détection d'une régurgitation tricuspide lors de l'échocardiographie Doppler du fœtus. Le premier diagnostic échographique prénatal de l'anomalie d'Ebstein a été réalisé à 18-19 semaines de gestation. Le pronostic de vie avec l'anomalie d'Ebstein est généralement favorable dans les cas où les enfants survivent au cours de la première année de vie sans traitement chirurgical. L'anomalie d'Ebstein n'est pas souvent associée à des aberrations chromosomiques et à des syndromes de malformations congénitales multiples. Des anomalies extracardiaques sont observées dans 25 %. Le résultat dans la période néonatale dépend de la gravité de la modification de la valve tricuspide. Les enfants atteints d'insuffisance valvulaire tricuspide sévère ont un pourcentage élevé de décès. Cliniquement, l'insuffisance de la valve tricuspide se manifeste par une augmentation de la cyanose, de l'acidose et des signes d'insuffisance cardiaque. Le traitement chirurgical est indiqué chez les patients présentant des symptômes graves de la maladie qui interfèrent avec la vie normale de l'enfant. L'opération consiste à fermer le septum
défaut, plasticité de la valve tricuspide et son mouvement vers un endroit typique. La mortalité hospitalière est de 6,3 %.
Tétralogie de Fallot- une malformation complexe, comprenant plusieurs anomalies de la structure du cœur : communication interventriculaire, dextroposition aortique, obstruction de l'artère pulmonaire de sortie et hypertrophie ventriculaire droite. Dans la structure générale des malformations cardiaques congénitales chez les naissances vivantes, la tétralogie de Fallot est de 4 à 11 %. Il est très difficile de diagnostiquer la tétrade de Fallot lors de l'étude d'un cœur fœtal à quatre chambres. Lorsqu'il est coupé à travers les sorties des artères principales, un VSD sous-aortique typique et une dextraposition aortique peuvent être observés. Un critère supplémentaire important est l'expansion et le déplacement de la racine aortique. La tétralogie de Fallot est un défaut de type bleu, c'est-à-dire chez les nouveau-nés, la cyanose prononcée est déterminée à l'âge de 6 semaines à 6 mois. La tétralogie de Fallot est une maladie cardiaque difficile à diagnostiquer qui passe souvent inaperçue lors du dépistage. examen échographique jusqu'à 22 semaines de gestation. Le plus souvent, ce défaut est diagnostiqué au troisième trimestre de la grossesse ou après la naissance. La tétralogie de Fallot ne nécessite pas de tactique de gestion spécifique. Si cette pathologie est détectée, un examen approfondi et une consultation prénatale sont nécessaires. Près de 30% des naissances vivantes avec tétrade de Fallot ont des anomalies extracardiaques combinées. Actuellement, plus de 30 syndromes de malformations multiples ont été décrits, dont la structure comprend la tétralogie de Fallot. L'examen prénatal pour la détection de la tétrade de Fallot doit inclure la détermination du caryotype et une étude détaillée de l'anatomie échographique du fœtus. Le pronostic vital dans la tétralogie de Fallot dépend en grande partie du degré d'obstruction des voies d'éjection du ventricule droit. Plus de 90% des patients ayant subi une correction complète de la tétrade de Fallot survivent jusqu'à l'âge adulte. A long terme, 80% des patients se sentent satisfaits et ont des paramètres fonctionnels normaux.
Transposition des grandes artères- une maladie cardiaque dans laquelle l'aorte, ou la majeure partie, sort du ventricule droit et l'artère pulmonaire du ventricule gauche. Il représente 5 à 7 % de toutes les malformations cardiaques congénitales. Habituellement non diagnostiqué dans la période prénatale lors du dépistage car l'étude du cœur fœtal est limitée à l'étude
seulement une coupe à quatre chambres. Pour identifier le défaut, la visualisation des vaisseaux principaux est nécessaire avec l'étude de leur emplacement les uns par rapport aux autres. Normalement, les artères principales se croisent et lors de la transposition, elles quittent les ventricules en parallèle: l'aorte - du ventricule droit, l'artère pulmonaire - de la gauche. La transposition des artères principales avec des cloisons interventriculaires et interauriculaires intactes n'est pas compatible avec la vie. Environ 8 % des naissances vivantes avec transposition des artères principales ont des anomalies extracardiaques associées. L'examen prénatal doit comprendre la détermination du caryotype et une étude détaillée de l'anatomie échographique du fœtus. La plupart des nouveau-nés présentant une transposition des artères principales et un septum interventriculaire intact présentent une cyanose prononcée dès les premiers jours de vie. Une correction chirurgicale doit être effectuée dès qu'un mélange inadéquat des flux sanguins est détecté. Le taux de mortalité des nouveau-nés avec ce type de traitement chirurgical est inférieur à 5-10%.
MALADIES DE LA POITRINE
hernie diaphragmatique congénitale- un défaut résultant d'un ralentissement du processus de fermeture du canal pleuropéritonéal. Avec ce défaut, il y a généralement un développement insuffisant de la partie postéro-latérale de la moitié gauche du diaphragme. Le manque de séparation entre la cavité abdominale et la poitrine entraîne le mouvement de l'estomac, de la rate, des intestins et même du foie dans la cavité thoracique, qui peut s'accompagner d'un déplacement médiastinal et provoquer une compression des poumons. En conséquence, une hypoplasie pulmonaire bilatérale de gravité variable se développe souvent. Le sous-développement des poumons entraîne une formation anormale de leur système vasculaire et une hypertension pulmonaire secondaire. La hernie diaphragmatique congénitale survient chez environ 1 nouveau-né sur 2400.
Il existe quatre principaux types de défauts : postérolatéral (hernie de Bochdalek), antérolatéral, sternal et hernie de Morgagni. Les hernies diaphragmatiques bilatérales représentent 1 % de tous les types de défauts. Mouvement du coeur vers la droite poitrine en combinaison avec une structure écho-négative (estomac) dans sa moitié gauche, il est le plus souvent diagnostiqué avec une hernie diaphragmatique du côté gauche.
Avec les hernies du côté droit, le cœur est généralement déplacé vers la gauche. Dans la poitrine, les intestins et le foie peuvent également être visualisés. Avec ce défaut, on note souvent un polyhydramnios. Des anomalies combinées sont observées chez 23 % des fœtus. Parmi eux, les malformations cardiaques congénitales prédominent, qui représentent 16 %. Le diagnostic du défaut peut être effectué dès 14 semaines de grossesse. La mortalité dans la hernie diaphragmatique congénitale est corrélée au moment de la détection de l'anomalie : seuls 33 % des nouveau-nés porteurs d'une anomalie survivent dans les cas où le diagnostic a été posé avant 25 semaines, et 67 % si la hernie a été détectée à une date ultérieure. Les anomalies diaphragmatiques ont généralement une origine multifactorielle, cependant, 12 % des cas sont associés à d'autres malformations ou font partie de syndromes chromosomiques et non chromosomiques. L'examen prénatal doit nécessairement inclure la détermination du caryotype fœtal et une échographie détaillée. Lorsque des anomalies combinées sont détectées diagnostic différentiel ne peut être réalisée qu'au cours d'une consultation avec la participation de généticiens, syndromologues, pédiatres. Il convient de conseiller aux parents de consulter un chirurgien pédiatrique pour discuter des caractéristiques des tactiques de traitement pendant la période néonatale, du pronostic vital et de la santé. L'évolution de la période néonatale dépend de la sévérité de l'hypoplasie pulmonaire et de la sévérité de l'hypertension. La taille de la hernie et le volume de tissu pulmonaire fonctionnel ont également un impact significatif sur l'issue de la période néonatale. Un développement anormal des poumons peut être prédit en présence de polyhydramnios, de dilatation de l'estomac, ainsi que du mouvement du foie fœtal dans la cavité thoracique. Selon la littérature, seulement 22 % des enfants diagnostiqués avant la naissance ont survécu. Même avec une hernie diaphragmatique congénitale isolée, seuls 40% survivent. La mort néonatale résulte généralement d'une hypertension pulmonaire et/ou d'une insuffisance respiratoire.
ANOMALIES DE FORMATION DES PAROIS DE LA CAVITÉ ABDOMINALE ET DE DÉVELOPPEMENT DU TRACTUS GASTRO-INTESTINAL
Omphalocèle (hernie ombilicale)(Fig. 32). Se produit à la suite d'un non-retour d'organes cavité abdominale de la cavité amniotique à travers l'anneau ombilical. L'omphalocèle peut inclure n'importe quel
 Riz. 32. Image échographique d'un omphalocèle (un sac herniaire est visualisé contenant des anses intestinales et un foie)
Riz. 32. Image échographique d'un omphalocèle (un sac herniaire est visualisé contenant des anses intestinales et un foie)
organes viscéraux. La taille de la formation herniaire est déterminée par son contenu.
Il est recouvert d'une membrane amniopéritonéale, le long de la surface latérale de laquelle passent les vaisseaux du cordon ombilical. La fréquence de l'omphalocèle est de 1 sur 3000 à 6000 nouveau-nés. Il existe des formes isolées et combinées d'omphalocèle. Cette pathologie dans 35 à 58% s'accompagne de trisomie, 47% - malformations cardiaques congénitales, 40% - malformations du système génito-urinaire, 39% - malformations du tube neural. Un retard de croissance intra-utérin est détecté dans 20% des cas.
Le diagnostic échographique prénatal repose sur la détection d'une formation ronde ou ovale remplie d'organes abdominaux et adjacente directement à la paroi abdominale antérieure. Le plus souvent, la composition du contenu herniaire comprend les anses intestinales et le foie. Le cordon ombilical est attaché directement au sac herniaire. Dans certains cas, un diagnostic prénatal peut être établi à la fin du premier trimestre de la grossesse, bien que dans la plupart des cas, un omphalocèle soit détecté au deuxième trimestre. Le pronostic dépend des anomalies qui l'accompagnent. Les pertes périnatales sont plus souvent associées à une maladie coronarienne, chromosomique
aberrations et prématurité. Le plus gros défaut est éliminé par une opération en une seule étape, avec une grande, des opérations en plusieurs étapes sont effectuées pour fermer le trou dans la paroi abdominale antérieure avec une membrane en silicone ou en téflon. La tactique obstétricale est déterminée par la période de détection du défaut, la présence d'anomalies combinées et de troubles chromosomiques. Si un défaut est détecté au début de la grossesse, il doit être interrompu. En cas de détection d'anomalies concomitantes incompatibles avec la vie, il est nécessaire d'interrompre la grossesse à tout moment. La méthode d'accouchement dépend de la viabilité du fœtus, car lors de l'accouchement avec un gros omphalocèle, le sac herniaire peut se rompre et une infection des organes internes du fœtus peut survenir.
Gastroschisis- défect de la paroi abdominale antérieure dans la région ombilicale avec éventération des anses intestinales couvertes d'exsudat inflammatoire. Le défaut est généralement situé à droite du nombril, les organes herniaires n'ont pas de membrane. La fréquence du gastroschisis est de 0,94 pour 10 000 nouveau-nés. La fréquence du défaut chez les femmes enceintes de moins de 20 ans est plus élevée et est de 7 pour 10 000 nouveau-nés.
Depuis la fin des années 70. 20ième siècle en Europe et aux États-Unis, la tendance à l'augmentation de la fréquence des naissances d'enfants atteints de gastroschisis se poursuit. Attribuez des formes isolées et combinées. Le gastroschisis isolé est plus fréquent et représente en moyenne 79 %. La forme combinée est détectée dans 10 à 30 % des cas et est le plus souvent une association de gastroschisis avec atrésie ou sténose intestinale. Entre autres anomalies, malformations congénitales du cœur et du système urinaire, syndrome ventre de pruneau, hydrocéphalie, bas et polyhydramnios.
L'anomalie survient sporadiquement, mais il existe des observations d'une maladie familiale avec un type de transmission autosomique dominant.
Le premier diagnostic échographique prénatal utilisant l'échographie transvaginale a été réalisé à 12 semaines de gestation. Dans la plupart des cas, le diagnostic est établi au deuxième trimestre de la grossesse, car dans les premiers stades (10-13 semaines), un diagnostic faussement positif est possible en raison de la présence d'une hernie intestinale physiologique chez le fœtus. Le diagnostic échographique prénatal du gastroschisis est généralement basé sur la visualisation des anses intestinales dans le liquide amniotique près de la paroi abdominale antérieure du fœtus. Parfois, en plus des anses intestinales, au-delà
d'autres organes peuvent être situés dans la cavité abdominale. La précision du diagnostic échographique du gastroschisis au cours des trimestres II et III de la grossesse varie de 70 à 95% et dépend de l'âge gestationnel, de la position du fœtus, de la taille du défaut et du nombre d'organes situés à l'extérieur de la paroi abdominale antérieure. .
Le pronostic global des nouveau-nés atteints de gastroschisis isolé est bon, plus de 90 % des nourrissons survivant. Lors de la prolongation de la grossesse, les tactiques de gestion au deuxième trimestre n'ont pas de particularités. En raison de la faible fréquence de l'association d'un gastroschisis isolé avec des anomalies chromosomiques, le caryotypage prénatal peut être évité. Au troisième trimestre de la grossesse, il est nécessaire de procéder à une évaluation dynamique de l'état fonctionnel du fœtus, car la fréquence de détresse dans le gastroschisis est assez élevée et un retard de croissance intra-utérin se forme dans 23 à 50% des cas.
Si un gastroschisis est détecté avant la viabilité du fœtus, un avortement doit être pratiqué. En cas de grossesse à terme, l'accouchement a lieu dans un établissement où des soins chirurgicaux peuvent être prodigués.
atrésie duodénale est la cause la plus fréquente d'occlusion de l'intestin grêle. La fréquence de l'anomalie est de 1 pour 10 000 naissances vivantes. L'étiologie est inconnue. La survenue d'un défaut sous l'influence de facteurs tératogènes est possible. Les observations familiales de l'atrésie pylo-duodénale à transmission autosomique récessive sont décrites. Chez 30 à 52 % des patients, l'anomalie est isolée, et chez 37 %, on retrouve des malformations du système squelettique : nombre anormal de côtes, agénésie du sacrum, pied de cheval, côtes cervicales bilatérales, absence bilatérale des premiers doigts , etc. Dans 2%, des anomalies combinées du tractus gastro-intestinal sont diagnostiquées: rotation incomplète de l'estomac, atrésie de l'œsophage, de l'iléon et de l'anus, transposition du foie. Chez 8 à 20% des patients, des malformations cardiaques congénitales sont détectées, dans environ 1/3 des cas, une atrésie duodénale est associée à une trisomie pour 21 paires de chromosomes. Les principales constatations échographiques prénatales dans l'atrésie duodénale sont l'hydramnios et le signe classique "double bulle" dans l'abdomen du fœtus. L'image de la "double bulle" apparaît à la suite de l'expansion d'une partie du duodénum et de l'estomac. La constriction entre ces formations est formée par la partie pylorique de l'estomac
ka et est d'une grande importance pour un diagnostic prénatal précis de ce défaut. Dans la grande majorité des cas, l'atrésie duodénale est diagnostiquée aux IIe et IIIe trimestres de la grossesse. En termes antérieurs, le diagnostic de ce défaut présente des difficultés importantes. Le premier diagnostic d'atrésie duodénale a été posé à 14 semaines.
Pour déterminer les tactiques obstétriques, une évaluation échographique détaillée de l'anatomie des organes internes du fœtus et de son caryotype est effectuée. Avant le début de la période de viabilité fœtale, une interruption de grossesse est indiquée. Si une anomalie isolée est détectée au troisième trimestre, une prolongation de grossesse est possible, suivie d'un accouchement au centre régional de périnatalité et d'une correction chirurgicale de la malformation.
Ascite isolée. L'ascite est l'accumulation de liquide dans la cavité péritonéale. La fréquence n'a pas été précisément établie. À l'examen échographique du fœtus, l'ascite se manifeste par la présence d'un espace écho-négatif d'une épaisseur de 5 mm ou plus dans la cavité abdominale. En période prénatale, l'ascite peut être isolée ou être l'un des signes d'une hydropisie d'origine non immunitaire. Outre l'ascite, l'hydropisie du fœtus se caractérise par la présence d'œdème sous-cutané, d'épanchements pleuraux et péricardiques, ainsi que par une augmentation de l'épaisseur du placenta de plus de 6 cm, un polyhydramnios et une hydrocèle.
L'ascite peut être associée à diverses anomalies structurelles. Un examen approfondi de tous les organes internes du fœtus est donc indiqué. Parmi les causes d'ascite isolée, il faut distinguer la péritonite méconiale et l'hépatite congénitale.
Jusqu'à présent, il n'y a pas eu de publications dans la littérature sur la détection de l'ascite isolée au premier trimestre de la grossesse. La plupart des observations de diagnostic précoce d'ascite surviennent au début du deuxième trimestre de la grossesse. Les anomalies chromosomiques sont l'une des causes les plus fréquentes d'hydropisie non immunitaire. Avec l'ascite isolée, les défauts chromosomiques sont moins fréquents, mais ils doivent être pris en compte comme arrière-plan possible pour le développement de cette pathologie. Lorsque l'ascite est détectée chez le fœtus, il faut d'abord exclure les anomalies combinées et les infections intra-utérines. L'évolution de l'ascite fœtale dépend de son étiologie. L'ascite isolée idiopathique a un pronostic favorable. Dans plus de 50% des observations, sa disparition spontanée est notée. La cause la plus fréquente d'ascite isolée est l'infection intra-utérine.
parvovirus B19. Lors de la prolongation de la grossesse, il est nécessaire d'effectuer une observation échographique dynamique, y compris une évaluation Doppler du flux sanguin dans le canal veineux. Avec des valeurs normales de débit sanguin dans le canal veineux chez les fœtus présentant une ascite, dans la plupart des cas, une issue périnatale favorable est notée. Avec une augmentation de l'ascite, certains auteurs recommandent une ponction thérapeutique, en particulier dans les cas où le processus progresse en fin de grossesse. Le but principal de la ponction est d'éviter les désagréments activité de travail et détresse respiratoire en période néonatale. Si une ascite isolée est détectée pendant la période prénatale et qu'une comorbidité incompatible avec la vie est exclue, l'enfant a besoin d'une surveillance dynamique attentive et d'un traitement symptomatique après l'accouchement.
Malformations des REINS ET DES VOIES URINAIRES
Agénésie rénale- absence complète des deux reins. L'apparition d'un défaut est due à une violation de la chaîne séquentielle des processus d'embryogenèse normale du pronéphros au métanéphros. La fréquence est en moyenne de 1:4500 nouveau-nés. On note qu'on le retrouve deux fois plus souvent chez les garçons. La triade pathognomonique des signes échographiques d'agénésie rénale chez le fœtus est représentée par l'absence de leur écho et de leur vessie, ainsi que par un oligohydramnios sévère. L'oligoamnios fait référence à des manifestations tardives et peut être détecté après la 16e à la 18e semaine de grossesse. Habituellement, l'agénésie rénale bilatérale s'accompagne d'une forme symétrique de syndrome de retard de croissance fœtale. L'agénésie rénale est le plus souvent sporadique, mais peut être associée à diverses anomalies des organes internes. Les conséquences directes de l'oligohydramnios sont l'hypoplasie pulmonaire, les malformations squelettiques et faciales, le syndrome de retard de croissance fœtale. L'agénésie rénale a été décrite dans plus de 140 syndromes de malformations congénitales multiples, d'anomalies chromosomiques et d'effets tératogènes. Une fois diagnostiqué, le caryotypage doit être effectué avant la naissance ou après la naissance pour exclure les anomalies chromosomiques. Dans toutes les observations d'agénésie rénale, un examen anatomopathologique complet est nécessaire. L'échographie est montrée
examens rénaux chez le plus proche parent. Avec la détection prénatale d'un défaut, l'interruption de grossesse à tout moment doit être recommandée. Si la famille décide de prolonger la grossesse, des tactiques obstétriques conservatrices sont indiquées.
Maladie polykystique des reins autosomique récessive (forme infantile). Elle se manifeste par une augmentation symétrique bilatérale des reins à la suite du remplacement du parenchyme par un canal collecteur secondairement élargi sans prolifération de tissu conjonctif. Elle va de la variante létale classique aux formes infantiles, juvéniles et même adultes. Dans la forme infantile, il existe une dilatation secondaire et une hyperplasie des canaux collecteurs normalement formés des reins. Les reins sont affectés de manière symétrique, tandis que les formations kystiques ont une taille de 1 à 2 mm. La fréquence est de 1,3-5,9:1000 nouveau-nés. Les principaux critères échographiques de malformation sont les reins hyperéchogènes hypertrophiés, l'absence d'écho vésical et l'oligohydramnios. L'augmentation de la taille des reins est parfois si importante qu'ils occupent une grande partie de la section transversale de l'abdomen fœtal. Une image échographique typique peut n'apparaître qu'au troisième trimestre de la grossesse. Le pronostic est défavorable. La mort vient d'une insuffisance rénale. La tactique obstétricale consiste à interrompre la grossesse à tout moment.
Maladie polykystique des reins de l'adulte(maladie autosomique dominante, polykystose hépatorénale de type adulte, syndrome de Potter de type III) se caractérise par le remplacement du parenchyme rénal par de nombreux kystes de différentes tailles, qui se forment en raison de l'expansion des canaux collecteurs et d'autres segments tubulaires du néphron. Les reins sont touchés des deux côtés et hypertrophiés, mais un processus unilatéral peut être la première manifestation de la maladie. Le foie est également impliqué dans le processus pathologique - une fibrose périportale se développe, qui a un caractère focal. L'étiologie de la maladie est inconnue, mais le type d'hérédité entraîne un risque de 50% de développer la maladie et son foyer génétique est situé sur la 16e paire de chromosomes. Le gène mutant est porté par une personne sur 1 000. La pénétration des gènes survient dans 100% des cas, cependant, l'évolution de la maladie peut varier de formes graves avec une issue fatale en période néonatale à des formes asymptomatiques détectées uniquement à l'autopsie.
Polykystose rénale(maladie polykystique, maladie rénale kystique, syndrome de Potter de type II, maladie rénale dysplasique) se caractérise par une dégénérescence kystique du parenchyme rénal due à l'expansion primaire des tubules rénaux. Dans la dysplasie rénale multikystique, l'uretère et le bassin sont le plus souvent atrésés ou absents. Le processus peut être bilatéral, unilatéral et segmentaire. Dans la dysplasie multikystique, le rein est généralement considérablement agrandi; la forme habituelle et le tissu normal sont absents. Le rein est représenté par de multiples kystes au contenu anéchoïque (Fig. 33).
 Riz. 33.Échogramme de reins fœtaux polykystiques bilatéraux (reins fortement hypertrophiés contenant plusieurs kystes de différents diamètres - indiqués par une flèche)
Riz. 33.Échogramme de reins fœtaux polykystiques bilatéraux (reins fortement hypertrophiés contenant plusieurs kystes de différents diamètres - indiqués par une flèche)
La taille des kystes varie dans une gamme assez large et dépend de la durée de la grossesse. Plus près du terme, le diamètre des kystes peut atteindre 3,5 à 4 cm.La vessie est généralement visualisée avec un processus unilatéral et n'est pas visualisée avec un processus bilatéral. Avec un processus bilatéral, un oligohydramnios est généralement noté. La maladie survient principalement de façon sporadique et peut être secondaire en association avec d'autres syndromes. obstétrique
tactique dans un processus bilatéral diagnostiqué dans les premiers stades, en raison d'un pronostic défavorable, consiste à interrompre la grossesse. Avec un processus unilatéral et un caryotype normal sans anomalies associées, un accouchement conventionnel est indiqué, suivi d'une consultation de l'enfant avec un spécialiste.
Dilatation des voies urinaires. Des anomalies du système génito-urinaire chez le fœtus, accompagnées de l'expansion des voies urinaires, peuvent être causées raisons diverses, dont le reflux vésico-urétéral, la pyélectasie idiopathique, les troubles obstructifs, etc. D'un point de vue clinique, il convient de distinguer la pyélectasie et l'uropathie obstructive en période prénatale.
Pyélectasie. La pyélectasie est caractérisée par une accumulation excessive de liquide et une expansion du bassinet du rein fœtal.
La pyélectasie est la découverte la plus fréquente à l'échographie fœtale. La fréquence de son développement n'a pas été établie, car cette pathologie est un phénomène sporadique. Après la naissance, les garçons en sont diagnostiqués 5 fois plus souvent. Chez 27% des enfants atteints d'hydronéphrose, un reflux vésico-urétéral, un doublement bilatéral des uretères, un mégauretère obstructif bilatéral, un rein controlatéral non fonctionnel et son agénésie sont détectés, chez 19% - des anomalies du développement de divers organes. Pour le diagnostic échographique prénatal de la pyélectasie, les reins fœtaux doivent être examinés à la fois en balayage transversal et longitudinal. La dilatation du pelvis rénal est jugée sur la base de sa taille antéro-postérieure lors d'un balayage transversal du rein. La plupart des chercheurs considèrent l'expansion pyélectasique du bassin rénal au cours du IIe trimestre de la grossesse de plus de 5 mm et au cours du IIIe trimestre - de plus de 8 mm. Avec l'expansion du bassin rénal du fœtus de plus de 10 mm, il est d'usage de parler d'hydronéphrose. La classification la plus courante de l'hydronéphrose chez le fœtus est la suivante :
Grade I (dilatation physiologique):
Bassin rénal : dimension antéro-postérieure<1 см;
Couche corticale : non modifiée.
Classe II :
Bassin rénal : 1,0-1,5 cm ;
Tasses : non visualisées ;
Couche corticale : non modifiée.
Degré III :
Bassin rénal : dimension antéropostérieure > 1,5 cm ;
Calices : légèrement dilatés ;
Couche corticale : non modifiée.
Classe IV :
Bassin rénal : dimension antéropostérieure > 1,5 cm ;
Calice : Modérément dilaté ;
Couche corticale : légèrement modifiée.
Degré V :
Bassin rénal : dimension antéropostérieure > 1,5 cm ;
Calices : fortement élargis ;
Couche corticale : atrophie.
L'expansion du bassin rénal du fœtus peut être observée avec diverses anomalies chromosomiques. La fréquence des anomalies chromosomiques chez les fœtus atteints de pyéloectasie est en moyenne de 8 %. Chez la plupart des fœtus présentant des anomalies chromosomiques, une combinaison de pyélectasie et d'autres anomalies du développement est détectée. Une pyélectasie modérément prononcée a un bon pronostic et la nécessité d'un traitement chirurgical après l'accouchement est assez rare. Dans la plupart des observations, on note une résolution spontanée d'une pyélectasie modérément sévère après la naissance d'un enfant.
La tactique obstétricale dépend du moment de l'apparition et de la durée de l'évolution du processus pathologique, ainsi que du degré d'altération de la fonction rénale. Un accouchement précoce est justifié par un oligohydramnios. Dans la période postnatale, une surveillance dynamique et une consultation avec un urologue pédiatrique sont présentées.
uropathie obstructive. L'obstruction des voies urinaires chez le fœtus peut être observée à n'importe quel niveau : obstruction élevée, obstruction au niveau de la fistule urétéro-pelvienne (PUR), obstruction au niveau moyen (uretère), obstruction au niveau de la jonction vésico-urétérale (VUR) , faible obstruction (urètre). L'URMS est la cause la plus fréquente d'uropathie obstructive chez le fœtus et représente en moyenne 50 % de toutes les anomalies urologiques congénitales. Les principales caractéristiques échographiques de l'OLMS comprennent la dilatation du bassinet du rein avec ou sans expansion des calices ; les uretères ne sont pas visualisés ; la vessie peut être de taille normale ou ne pas être visualisée dans certains cas. Les tactiques en OLMS doivent être attendues. La pose d'un shunt vésico-amniotique n'est pas indiquée. Au cri ultrasonique
Les OPMS chez le fœtus comprennent la dilatation de l'uretère et la pyélectasie. La vessie est généralement de taille normale. Les tactiques de direction sont similaires à celles de l'OLMS. La cause la plus fréquente d'obstruction basse est les valves urétrales postérieures. En cas d'obstruction sévère, on observe un oligohydramnios, entraînant une hypoplasie pulmonaire, des déformations des structures faciales et des membres, une fibrose et une dysplasie du parenchyme rénal. L'image échographique est caractérisée par la présence d'un urètre dilaté à proximité du site d'obstruction, une expansion prononcée de la vessie. La prise en charge prénatale de la faible obstruction dépend de la durée de la grossesse, de la présence d'oligohydramnios et d'anomalies associées, et de l'état fonctionnel des reins. Avec une pyélectasie modérément sévère et non progressive, des tactiques conservatrices doivent être suivies. Avec la progression des troubles obstructifs, l'accouchement avec correction chirurgicale éventuelle du défaut est justifié pour prévenir les troubles rénaux graves chez le fœtus. En cas de grossesse prématurée chez les fœtus atteints d'uropathie obstructive sévère, une correction chirurgicale intra-utérine du défaut peut être effectuée.
DÉFAUTS OSSEUX
Parmi les malformations congénitales du système squelettique, l'amélie (aplasie de tous les membres) est la plus fréquente ; phocomélie (sous-développement des membres proximaux, alors que les mains et les pieds sont directement reliés au corps); aplasie de l'un des os de la jambe ou de l'avant-bras inférieur; polydactylie (une augmentation du nombre de doigts sur un membre); syndactylie (réduction du nombre de doigts due à la fusion des tissus mous ou du tissu osseux des doigts adjacents) ; réglage d'arrêt anormal ; l'ostéochondrodysplasie, caractérisée par des anomalies de la croissance et du développement du cartilage et/ou des os (achondrogenèse, achondroplasie, dysplasie thanatoforme, ostéogenèse imparfaite, hypophosphatasie, etc.).
Le plus important est le diagnostic des malformations incompatibles avec la vie. De nombreuses formes de dysplasie squelettique sont associées à une hypoplasie pulmonaire due à la petite taille de la poitrine due au sous-développement des côtes. Le développement d'une insuffisance pulmonaire dans ce cas peut être la cause du décès d'enfants dans les premières heures de la vie extra-utérine.
Achondroplasie est l'une des dysplasies squelettiques non létales les plus fréquentes et est causée par une nouvelle mutation dans 90% des cas. L'achondroplasie est une ostéochondroplasie avec des défauts des os longs et/ou du squelette axial. La fréquence est de 0,24-5:10 000 naissances. Le ratio de fœtus mâles et femelles est de 1:1. Le raccourcissement des os dans l'achondroplasie peut ne pas apparaître chez le fœtus avant 24 semaines de gestation. Les signes échographiques classiques incluent des membres courts (inférieurs au 5e centile), une petite poitrine, une macrocéphalie et un nez en selle. L'espérance de vie avec l'achondroplasie dépend principalement du moment où la petite taille de la poitrine n'entraîne pas de problèmes respiratoires graves. Développement intellectuel avec un défect, c'est normal, mais il existe un risque élevé de troubles neurologiques, en particulier, une compression de la moelle épinière au niveau du foramen magnum, ce qui peut limiter exercice physique. La macrocéphalie peut être le résultat d'une hydrocéphalie légère due à la petite taille du foramen magnum. L'achondroplasie est un type bien étudié et courant de nanisme congénital chez les nouveau-nés. Problèmes graves, ils peuvent avoir une apnée centrale et obstructive du sommeil. À l'âge de 6-7 ans, on note souvent des infections chroniques récurrentes de l'oreille moyenne. Au début enfance il y a aussi souvent une courbure des membres inférieurs qui, dans des conditions sévères, nécessite une correction chirurgicale. Habituellement, la taille des adultes atteints d'achondroplasie varie de 106 à 142 cm.
Le concept de teragotèse
Le nom de la science de la tératologie provient du mot "teras", qui en grec signifie "monstre". La tératogenèse se traduit littéralement par la reproduction de monstres. À l'heure actuelle, ce terme en est venu à être compris comme divers troubles chez les nouveau-nés de nature fonctionnelle, qui comprenaient également un retard de croissance intra-utérin généralisé et d'autres changements de comportement en résultant. Jusque dans les années 1950, on ne savait rien de la tératogenèse et on croyait que les changements dans les gènes étaient à l'origine de la plupart des anomalies congénitales.
malformations congénitales: classification
Selon la fréquence de leur apparition, toutes les anomalies intra-utérines sont divisées en trois groupes:
- Les malformations courantes sont considérées comme courantes si elles surviennent dans une population avec une fréquence supérieure à 1 cas pour mille nouveau-nés ;
- Survenant modérément (leur fréquence est de 0,1 à 0,99 cas pour mille nouveau-nés) ;
- Malformations congénitales rares (moins de 0,01 pour mille enfants).
En fonction de sa distribution dans l'organisme CM enfant distinguer:
- nature isolée (en règle générale, un organe est affecté);
- systémique (malformation du système organique);
- Multiple (plusieurs systèmes sont concernés).
défaut de naissance par gravité manifestations cliniques et la prévision pour la vie se produit:
- Mortel, qui entraîne la mort de l'enfant. La fréquence de ces malformations congénitales est en moyenne d'environ 0,5%, jusqu'à 85% des enfants atteints de ces anomalies ne survivent qu'à la fin de la première année de vie;
- Modéré-sévère, dans laquelle une intervention chirurgicale est nécessaire pour le corriger (jusqu'à 2,5%);
- MAP (anomalie mineure du développement), qui ne nécessite pas de chirurgie et ne limite pas la vie de l'enfant (environ 4%).
Selon le temps d'exposition au facteur négatif, VPR est divisé en:

La pathogenèse des anomalies congénitales
Les mécanismes pathogéniques de l'apparition des défauts sont actuellement bien compris. Si l'embryon est endommagé avant l'introduction dans la muqueuse utérine, il se produit soit sa mort (en cas de modifications irréversibles des cellules), soit sa récupération (en cas de lésions réversibles). Au fur et à mesure que l'embryon se développe, les mécanismes de réparation cellulaire cessent de fonctionner et toute violation entraînera la formation d'un défaut. Le contrôle génétique de l'embryogenèse peut être altéré à la suite d'une exposition à divers facteurs agressifs externes (tératogènes).
Les principaux mécanismes de tératogenèse chez le fœtus au niveau cellulaire seront: les violations de la division cellulaire (il y a un sous-développement de l'organe), leur mouvement (l'organe sera situé au mauvais endroit) et la différenciation (l'absence d'un organe ou système d'organes). Au niveau tissulaire, les processus tératogènes seront: mort prématurée des cellules, retard de leur décomposition et de leur résorption, violation du processus de collage, entraînant des défauts tels que la fermeture de l'ouverture naturelle, une fistule, un défaut dans les tissus, etc.
Quels sont les principaux facteurs de risque pour lesquels défaut de naissance beaucoup plus courant ?
Les principaux facteurs contributifs sont :
- grossesse non planifiée;
- Âge mère (plus de 35 ans);
- Contrôle médical insuffisant avant la conception ;
- L'incidence des infections virales ;
- Prendre des médicaments qui ont un effet prononcé influence négative sur le fœtus;
- Boire de l'alcool et fumer;
- l'usage de drogues;
- Malnutrition;
- La présence de risques professionnels ;
- Financement insuffisant des soins de santé dans de nombreux pays.
La présence de quelles conditions pathologiques est une indication pour la prophylaxie prénatale des malformations congénitales ?
 Pour que l'enfant à naître n'ait pas malformations congénitales, une femme doit se préparer à l'avance à la conception et à la grossesse en présence des facteurs suivants:
Pour que l'enfant à naître n'ait pas malformations congénitales, une femme doit se préparer à l'avance à la conception et à la grossesse en présence des facteurs suivants:
- Diabète sucré et autres maladies du système endocrinien et du métabolisme ;
- Fausses couches spontanées antérieures et mort fœtale intra-utérine ;
- La présence d'une prédisposition héréditaire aux malformations;
- La naissance d'enfants précédents avec un retard de croissance intra-utérin ou prématurément à l'âge gestationnel ;
- La présence de diverses maladies chroniques (hypertension artérielle, épilepsie, asthme bronchique, etc.);
- surpoids et obésité;
- L'usage de drogues pendant une longue période;
- Maladies infectieuses (surtout toxoplasmose et rubéole).
Comment prévenir les MCV ?
Le schéma des mesures prises pour prévenir d'éventuels défauts comprend:

Qu'avez-vous besoin de savoir?
Eduardo Castillo, un généticien brésilien, a formulé dix commandements de base pour la prévention des malformations congénitales des futurs enfants. Ils comprennent les éléments suivants :
- Une femme doit se rappeler que si elle est capable de tomber enceinte, elle peut être enceinte à tout moment ;
- Vous devez compléter votre famille quand vous êtes encore jeune ;
- Il est nécessaire dans le bon ordre de passer, si nécessaire, le contrôle prénatal;
- Il est conseillé de vacciner contre la rubéole avant la conception ;
- Il est nécessaire d'exclure l'utilisation de drogues, sauf pour les plus nécessaires pour vous;
- Ne buvez pas d'alcool et ne fumez pas;
- il est également conseillé d'éviter les zones fumeurs;
- Assurez-vous de bien manger et pleinement, de préférence des légumes et des fruits;
- Connaître les risques de grossesse dans votre milieu de travail;
- En cas de doute, demandez des réponses à votre médecin pour toutes les questions.
Photo : Alexander Anatolievich Kryukov, orthopédiste, MD
Ainsi, on peut conclure que la plupart des anomalies congénitales chez le fœtus résultent d'un développement altéré de l'œuf fécondé. Une telle violation peut survenir à tout moment après la conception. Il a été prouvé que plus tôt cela se produit fausse-couche plus le changement est important. Au cours des trois premiers mois de gestation, environ 75 % des avortements spontanés s'expliquent par la présence de diverses mutations dans les gènes et les chromosomes. L'acide folique a la capacité d'améliorer les propriétés réparatrices du fœtus et de le protéger des dommages, il est donc recommandé à toutes les femmes à risque de survenue de malformations congénitales.
Les malformations congénitales du fœtus (CM) sont peut-être la complication la plus dangereuse de la grossesse, entraînant une invalidité et une mortalité infantiles.
La naissance d'un enfant atteint de malformations congénitales du développement est toujours un grand traumatisme pour tout parent. Les statistiques à cet égard ne sont pas rassurantes : en Russie, la fréquence des malformations congénitales atteint 5 à 6 cas pour 1000 enfants.
Malheureusement, il n'est pas possible de prédire ces pathologies avant la grossesse. Un enfant atteint de malformations congénitales peut apparaître dans n'importe quelle famille, indépendamment de la présence ou de l'absence de mauvaises habitudes, de mode de vie ou de richesse matérielle.
Quels sont les troubles du développement du fœtus pendant la grossesse ?
Toutes les anomalies du développement du fœtus pendant la grossesse peuvent être divisées pour plusieurs types:
1. héréditaire
Les maladies héréditaires sont le résultat de mutations génétiques. Une mutation est une modification des propriétés héréditaires d'un organisme due à des réarrangements dans les structures responsables du stockage et de la transmission de l'information génétique. Ceux-ci incluent le syndrome de Down, le syndrome de Patau, etc.
2. Congénital
Les anomalies congénitales sont des maladies acquises dans l'utérus suite à une exposition à facteurs externes(et oligo-éléments, traumatisme pendant la grossesse, etc.). Ils peuvent affecter presque tous les organes. Les malformations congénitales du fœtus comprennent les malformations cardiaques, le sous-développement du cerveau, les déformations maxillo-faciales, etc.
3. Multifactoriel (facteur combiné)
La division des anomalies du développement fœtal en types est plutôt arbitraire, car dans la grande majorité des cas, les retards de développement sont une combinaison de facteurs héréditaires et congénitaux.
Classification des malformations fœtales
Les malformations les plus courantes du développement intra-utérin du fœtus:
- Aplasie (absence de tout organe);
- Dystopie (emplacement de l'organe dans un endroit inhabituel pour lui);
- Ectopie (déplacement d'un organe vers l'extérieur ou dans une cavité corporelle adjacente);
- Hypotrophie, hypoplasie (perte de poids du fœtus, sous-développement);
- Hypertrophie, hyperplasie (augmentation de la taille de n'importe quel organe);
- Atrésie (infection des ouvertures naturelles);
- Fusion d'organes appariés ;
- Sténose (rétrécissement des canaux et ouvertures des organes fœtaux);
- Gigantisme (augmentation de la taille du corps et des organes internes du fœtus);
- Dyschronie (accélération ou inhibition du développement des processus).
Il convient de noter que la gravité des pathologies peut être complètement différente. Cela dépend de la localisation des dommages génétiques, ainsi que de la durée et de l'intensité de l'effet toxique sur le fœtus. Il n'y a pas de relation claire entre eux.
Une femme qui a été exposée à des effets toxiques pendant sa grossesse peut accoucher tout à fait enfant en bonne santé. Dans le même temps, le risque de retard de développement chez la future progéniture de ce fœtus demeure, en raison de dommages génétiques avec l'absence de manifestations cliniques.
Causes des malformations fœtales
La problématique de l'étude des pathologies du développement fœtal est très diverse. Ce sujet est traité par des spécialistes de différents niveaux et directions - génétique, embryologistes, néonatologistes, spécialistes du diagnostic prénatal.
La cause des pathologies héréditaires est une mutation génétique. Divers effets indésirables sur les organes du fœtus pendant la grossesse, en particulier pendant les périodes critiques de son développement, entraînent l'apparition d'anomalies congénitales. Les facteurs qui causent CM sont appelés tératogènes.
Les facteurs tératogènes les plus étudiés:
- médicaments (prise de médicaments interdits pendant la grossesse ou pendant une certaine période de grossesse) ;
- infectieux (rougeole, varicelle, transmis de la mère au fœtus);
- rayonnement ionisant (rayons X, rayonnement radioactif);
- facteur alcool (une grande quantité d'alcool prise par une femme enceinte peut entraîner un syndrome alcoolique sévère chez le fœtus, incompatible avec la vie);
- facteur nicotine (fumer pendant la grossesse peut provoquer un retard dans le développement de l'enfant);
- toxiques et chimiques (les femmes travaillant dans des industries dangereuses doivent éviter tout contact avec des substances chimiques et toxiques agressives quelques mois avant la grossesse et pendant toute sa durée afin d'éviter un effet tératogène chez le fœtus);
- manque de vitamines et de microéléments (manque d'acide folique et d'acides polyinsaturés oméga-3, de protéines, d'iode, le manque d'une alimentation équilibrée peut entraîner un retard dans le développement du fœtus, des troubles cérébraux).
Souvent, la prédisposition héréditaire joue un rôle important dans l'apparition du CM fœtal. Si les parents ou les proches parents de l'enfant avaient des malformations congénitales, le risque de donner naissance à un enfant avec les mêmes défauts augmente plusieurs fois.
Périodes critiques du développement fœtal
Le développement intra-utérin du fœtus dure en moyenne 38 à 42 semaines. Pendant tout ce temps, le fœtus est bien protégé des agressions extérieures par la barrière placentaire et le système immunitaire de la mère. Mais il y a 3 périodes critiques pendant lesquelles il est très vulnérable aux agents nocifs. Par conséquent, à ce moment, une femme enceinte doit surtout prendre soin d'elle-même.
La première période critique survient environ 7 à 8 jours après la fécondation, lorsque l'embryon passe par la phase d'implantation dans l'utérus. La prochaine période dangereuse est de 3 à 7 et de 9 à 12 semaines de grossesse, lorsque le placenta est formé. La maladie, l'exposition aux produits chimiques ou aux rayonnements d'une femme enceinte pendant ces périodes peut entraîner des malformations intra-utérines du fœtus.
La troisième période critique de la grossesse est de 18 à 22 semaines, lorsque la pose des connexions neuronales du cerveau se produit et que le système hématopoïétique commence son travail. Il y a un retard associé à cela développement mental fœtus.
Facteurs de risque d'anomalies fœtales
Facteurs de risque maternels pour CM :
- âge supérieur à 35 ans - retard de croissance intra-utérin, troubles génétiques;
- jusqu'à 16 ans - prématurité, manque de vitamines et de minéraux;
- statut social bas - infections, hypoxie fœtale, prématurité, retard de croissance intra-utérin;
- manque d'acide folique - malformations congénitales du système nerveux;
- alcool, drogues et tabagisme - retard de croissance intra-utérin, syndrome de mort subite, syndrome d'alcoolisation fœtale ;
- infections (varicelle, rubéole, infections herpétiques, toxoplasmose) - malformations congénitales, retard de croissance intra-utérin, pneumonie, encéphalopathie;
- hypertension artérielle - retard de croissance intra-utérin, asphyxie;
- polyhydramnios - malformations congénitales du système nerveux central, pathologies du tractus gastro-intestinal et des reins;
- maladies thyroïdiennes - hypothyroïdie, thyrotoxicose, goitre;
- maladie rénale - retard de croissance intra-utérin, néphropathie, mortinatalité;
- maladies des poumons et du cœur - malformations cardiaques congénitales, retard de croissance intra-utérin, prématurité;
- anémie - retard de croissance intra-utérin, mortinatalité;
- saignement - anémie, prématurité, mortinaissance
Facteurs de risque de malformations congénitales du fœtus :
- anomalies de la présentation fœtale - hémorragie, malformations congénitales, traumatismes ;
- grossesse multiple - transfusion fœto-fœtale, asphyxie, prématurité ;
- retard de croissance intra-utérin - mortinaissance, malformations congénitales, asphyxie,
Facteurs de risque lors de l'accouchement : - naissance prématurée - lourde de développement de l'asphyxie;
- accouchement tardif (accouchement retardé de 2 semaines ou plus) - le développement d'une asphyxie ou d'une mortinaissance est possible ;
- accouchement prolongé - asphyxie, mortinatalité;
- prolapsus du cordon ombilical - asphyxie.
Anomalies dans le développement du placenta :
- petit placenta - retard de croissance intra-utérin;
- gros placenta - développement de l'hydropisie du fœtus, insuffisance cardiaque;
- décollement prématuré du placenta - une perte de sang importante est possible, le développement d'une anémie;
- placenta praevia - lourd de pertes de sang et de développement d'anémie.
Diagnostic des malformations fœtales
Diagnostic prénatal des anomalies fœtales et pathologies génétiques- le processus est très complexe. L'une des étapes de ce diagnostic consiste en des examens de dépistage prescrits à une femme enceinte pour une période de 10-12, 20-22 et 30-32 semaines (dans chacun des trimestres). Cette analyse est un test sanguin pour les marqueurs sériques biochimiques de la pathologie chromosomique (malformations).
Cela permettra d'obtenir une hypothèse sur la présence ou l'absence d'anomalies chromosomiques chez le fœtus, et l'échographie en tant que méthode de diagnostic supplémentaire montrera s'il existe des écarts dans Développement physique fœtus. L'échographie doit être effectuée par un spécialiste hautement qualifié et sur un équipement de haute qualité. Les résultats de chaque étude sont évalués conjointement, sans séparation les uns des autres.
Le dépistage ne garantit pas une pathologie à 100%, il permet uniquement d'identifier un groupe à haut risque parmi les femmes enceintes. Il s'agit d'une mesure importante et nécessaire et, malgré le caractère volontaire, la plupart des femmes enceintes le comprennent. Il n'est pas rare que les spécialistes aient du mal à répondre à la question de la présence d'anomalies génétiques chez le fœtus. Ensuite, selon le trimestre de la grossesse, la patiente se voit prescrire méthodes de recherche invasives :
- (étude des villosités choriales)
Elle se fait au 1er trimestre de la grossesse (11-12 semaines) et permet d'identifier des anomalies génétiques dans le développement du fœtus.
- amniocentèse (examen du liquide anatomique dans lequel se trouve le fœtus)
Au 1er trimestre, cette analyse révèle une hyperplasie du cortex surrénalien, au 2ème - maladies du SNC, pathologies chromosomiques.
- placentocentèse (examen des particules placentaires)
Elle est pratiquée de 12 à 22 semaines de grossesse pour détecter des pathologies génétiques.
- (prélèvement sanguin du cordon ombilical du fœtus)
Permet d'identifier la susceptibilité du fœtus aux maladies génétiques ou infectieuses.
Les femmes enceintes sont envoyées pour une consultation obligatoire avec un généticien :
- dont l'âge dépasse 35 ans;
- avoir un enfant ou des enfants atteints de troubles génétiques ;
- qui avaient des antécédents de fausses couches, de grossesses non évolutives, de mortinatalité ;
- dans la famille de qui il y a des parents atteints du syndrome de Down et d'autres anomalies chromosomiques ;
- récupéré de maladies virales au 1er trimestre de la grossesse;
- prendre des médicaments interdits pendant la grossesse;
- exposé aux radiations.
Pour le diagnostic des pathologies fœtales après la naissance, les méthodes de recherche suivantes : analyses de sang, d'urine et d'autres fluides biologiques, rayons X, imagerie par résonance magnétique et informatisée, échographie, angiographie, broncho et gastroscopie, autres méthodes immunitaires et moléculaires ...
Indications pour l'interruption de grossesse
Toute détection de malformations fœtales implique une proposition d'interruption de grossesse pour des raisons dites médicales. Si une femme refuse et décide de garder l'enfant, elle est placée sous contrôle spécial et la grossesse est surveillée plus attentivement.
Mais femme enceinte il faut comprendre que non seulement ses sentiments et ses expériences sont importants ici, mais aussi le fait que les enfants nés avec de graves malformations et pathologies s'avèrent souvent non viables ou restent gravement handicapés à vie, ce qui, bien sûr, est très difficile pour tout famille.
Il existe d'autres indications à l'avortement:
- néoplasmes malins (la grossesse avec cancer est contre-indiquée);
- maladies du système cardiovasculaire (malformations cardiaques, thrombose veineuse profonde, thromboembolie);
- maladies neurologiques ( sclérose en plaques, myasthénie grave);
- maladies infectieuses (, sous une forme active, à un stade aigu et sévère,);
- maladies du sang et des organes hématopoïétiques (hémoglobinopathie, anémie aplasique, leucémie);
- maladies oculaires (maladies du nerf optique et de la rétine);
- maladie rénale (lithiase urinaire aiguë et avec gros calculs, aiguë);
- maladies diffuses du tissu conjonctif;
- troubles endocriniens (, thyrotoxicose, hypothyroïdie non compensée dans les formes sévères) ;
- certaines maladies gynécologiques;
- indications obstétricales (insensibles au traitement et sévères, accompagnées de vomissements sévères, maladie trophoblastique gestationnelle, maladies héréditaires sévères détectées pendant la grossesse, etc.)
L'avortement pour raisons médicales n'est pratiqué qu'avec le consentement de la patiente.
Prévention des malformations congénitales du fœtus
La principale mesure visant à prévenir la survenue de malformations congénitales du fœtus est la planification de la grossesse. Non seulement le succès de la conception, mais aussi le processus de réalisation d'une grossesse, un accouchement rapide et correct et la santé de la mère et de l'enfant à l'avenir peuvent dépendre d'une préparation de haute qualité.
Avant de planifier une grossesse, il est nécessaire de subir une série d'examens : faire des tests de dépistage (MST), VIH, hépatite, syphilis, vérifier la coagulation du sang, le statut hormonal, assainir la cavité buccale, faire une échographie des organes pelviens pour exclure les inflammations maladies et néoplasmes, consultez un thérapeute pour identifier toutes les maladies chroniques possibles, idéalement les deux parents devraient être testés génétiquement.
Le point clé de la prévention des anomalies congénitales du fœtus est le maintien mode de vie sain la vie, en abandonnant les mauvaises habitudes, une alimentation équilibrée et nutritive, en éliminant l'impact sur votre corps de tout facteur négatif et nocif. Pendant la grossesse, il est important de traiter toutes les maladies possibles en temps opportun et de suivre les instructions de l'obstétricien-gynécologue.
Traitement des malformations congénitales du fœtus
Les méthodes de traitement des malformations congénitales du fœtus varient considérablement selon la nature et la gravité de l'anomalie. Les statistiques sur cette question ne sont malheureusement pas encourageantes. Un quart des enfants anomalies congénitales meurt dans la première année de vie.
Un autre 25% peut vivre assez longtemps, tout en ayant des troubles physiques et mentaux intraitables ou difficiles à traiter. Et seuls 5 % des enfants nés avec des malformations congénitales peuvent être traités, le plus souvent chirurgicalement. Dans certains cas, un traitement conservateur aide. Parfois, les malformations ne deviennent perceptibles qu'en vieillissant, certaines sont complètement asymptomatiques.
Les malformations congénitales (MC) sont les complications les plus dangereuses de la grossesse. En raison de malformations congénitales du fœtus, l'enfant peut devenir handicapé et, dans les cas graves, entraîner la mort. Pour inclure les malformations congénitales du système nerveux central fœtal :
- anencéphalie (pas de cerveau)
- spina bifida (forme ouverte de moelle épinière herniée);
- MVS VFR du fœtus ;
- modifications pathologiques du système cardiovasculaire (maladies cardiaques);
- défauts des membres - atrésie (les membres sont absents);
- déformations maxillo-faciales - fente palatine ou fente labiale.
Causes de CM fœtale
Le développement de malformations fœtales peut survenir sous l'influence de nombreux facteurs. La plupart d'entre eux restent inexpliqués. Les signes étiologiques de tous les CM fœtaux sont divisés en :
- héréditaire - déviations dans les ensembles chromosomiques des parents;
- tératogène - le fœtus ou l'embryon a été endommagé par l'action de pesticides, d'infections, de médicaments, etc. ;
- multifactoriel - un effet conjoint sur le fœtus de facteurs génétiques et environnementaux, qui séparément ne peuvent pas être la cause du défaut.
Il est prouvé que la pollution de la biosphère est la cause de soixante-dix pour cent des cas de maladies, soixante pour cent des cas de développement avec pathologies et plus de cinquante pour cent des décès chez les enfants. La naissance d'enfants avec un développement anormal ultérieur est associée à activité professionnelle: stress émotionnel, exposition à des basses températures ou poussière, contact avec le produit industrie chimique et les sels de métaux lourds.
Un risque plus élevé de malformations congénitales chez le fœtus chez les femmes présentant une obésité importante.  Cela peut provoquer des anomalies du tube neural. Mais pas seulement surpoids enceinte, et une forte perte de poids sur premières dates grossesse.
Cela peut provoquer des anomalies du tube neural. Mais pas seulement surpoids enceinte, et une forte perte de poids sur premières dates grossesse.
Grossesse après fœtus congénital
Il est possible de planifier une grossesse après une interruption due au développement de malformations congénitales chez le fœtus dès six mois après la précédente. Dans certains cas, il est conseillé au couple d'attendre un an. Dans le processus de planification, les futurs parents passent par une série d'études, selon les résultats desquelles le médecin recommande quand il est possible de concevoir un enfant. En préparation de la prochaine grossesse, le couple doit mener une vie saine, éviter l'influence de facteurs négatifs, prendre des vitamines et d'autres substances utiles afin de renforcer son corps.
Les malformations congénitales du fœtus (CM) sont peut-être la complication la plus dangereuse de la grossesse, entraînant une invalidité et une mortalité infantiles.
La naissance d'un enfant atteint de malformations congénitales du développement est toujours un grand traumatisme pour tout parent. Les statistiques à cet égard ne sont pas rassurantes : en Russie, la fréquence des malformations congénitales atteint 5 à 6 cas pour 1000 enfants.
Malheureusement, il n'est pas possible de prédire ces pathologies avant la grossesse. Un enfant atteint de malformations congénitales peut apparaître dans n'importe quelle famille, indépendamment de la présence ou de l'absence de mauvaises habitudes, de mode de vie ou de richesse matérielle.
Quels sont les troubles du développement du fœtus pendant la grossesse ?
Toutes les anomalies du développement du fœtus pendant la grossesse peuvent être divisées pour plusieurs types:
1. héréditaire
Les maladies héréditaires sont le résultat de mutations génétiques. Une mutation est une modification des propriétés héréditaires d'un organisme due à des réarrangements dans les structures responsables du stockage et de la transmission de l'information génétique. Ceux-ci incluent le syndrome de Down, le syndrome de Patau, etc.
2. Congénital
Les anomalies congénitales sont des maladies acquises dans l'utérus sous l'influence de facteurs externes (et oligo-éléments, traumatismes pendant la grossesse, etc.). Ils peuvent affecter presque tous les organes. Les malformations congénitales du fœtus comprennent les malformations cardiaques, le sous-développement du cerveau, les déformations maxillo-faciales, etc.
3. Multifactoriel (facteur combiné)
La division des anomalies du développement fœtal en types est plutôt arbitraire, car dans la grande majorité des cas, les retards de développement sont une combinaison de facteurs héréditaires et congénitaux.
Classification des malformations fœtales
Les malformations les plus courantes du développement intra-utérin du fœtus:
- Aplasie (absence de tout organe);
- Dystopie (emplacement de l'organe dans un endroit inhabituel pour lui);
- Ectopie (déplacement d'un organe vers l'extérieur ou dans une cavité corporelle adjacente);
- Hypotrophie, hypoplasie (perte de poids du fœtus, sous-développement);
- Hypertrophie, hyperplasie (augmentation de la taille de n'importe quel organe);
- Atrésie (infection des ouvertures naturelles);
- Fusion d'organes appariés ;
- Sténose (rétrécissement des canaux et ouvertures des organes fœtaux);
- Gigantisme (augmentation de la taille du corps et des organes internes du fœtus);
- Dyschronie (accélération ou inhibition du développement des processus).
Il convient de noter que la gravité des pathologies peut être complètement différente. Cela dépend de la localisation des dommages génétiques, ainsi que de la durée et de l'intensité de l'effet toxique sur le fœtus. Il n'y a pas de relation claire entre eux.
Une femme qui a été exposée à des effets toxiques pendant sa grossesse peut donner naissance à un bébé en parfaite santé. Dans le même temps, le risque de retard de développement chez la future progéniture de ce fœtus demeure, en raison de dommages génétiques avec l'absence de manifestations cliniques.
Causes des malformations fœtales
La problématique de l'étude des pathologies du développement fœtal est très diverse. Ce sujet est traité par des spécialistes de différents niveaux et directions - génétique, embryologistes, néonatologistes, spécialistes du diagnostic prénatal.
La cause des pathologies héréditaires est une mutation génétique. Divers effets indésirables sur les organes du fœtus pendant la grossesse, en particulier pendant les périodes critiques de son développement, entraînent l'apparition d'anomalies congénitales. Les facteurs qui causent CM sont appelés tératogènes.
Les facteurs tératogènes les plus étudiés:
- médicaments (prise de médicaments interdits pendant la grossesse ou pendant une certaine période de grossesse) ;
- infectieux (rougeole, varicelle, transmis de la mère au fœtus);
- rayonnement ionisant (rayons X, rayonnement radioactif);
- facteur alcool (une grande quantité d'alcool prise par une femme enceinte peut entraîner un syndrome alcoolique sévère chez le fœtus, incompatible avec la vie);
- facteur nicotine (fumer pendant la grossesse peut provoquer un retard dans le développement de l'enfant);
- toxiques et chimiques (les femmes travaillant dans des industries dangereuses doivent éviter tout contact avec des substances chimiques et toxiques agressives quelques mois avant la grossesse et pendant toute sa durée afin d'éviter un effet tératogène chez le fœtus);
- manque de vitamines et de microéléments (manque d'acide folique et d'acides polyinsaturés oméga-3, de protéines, d'iode, le manque d'une alimentation équilibrée peut entraîner un retard dans le développement du fœtus, des troubles cérébraux).
Souvent, la prédisposition héréditaire joue un rôle important dans l'apparition du CM fœtal. Si les parents ou les proches parents de l'enfant avaient des malformations congénitales, le risque de donner naissance à un enfant avec les mêmes défauts augmente plusieurs fois.
Périodes critiques du développement fœtal
Le développement intra-utérin du fœtus dure en moyenne 38 à 42 semaines. Pendant tout ce temps, le fœtus est bien protégé des agressions extérieures par la barrière placentaire et le système immunitaire de la mère. Mais il y a 3 périodes critiques pendant lesquelles il est très vulnérable aux agents nocifs. Par conséquent, à ce moment, une femme enceinte doit surtout prendre soin d'elle-même.
La première période critique survient environ 7 à 8 jours après la fécondation, lorsque l'embryon passe par la phase d'implantation dans l'utérus. La prochaine période dangereuse est de 3 à 7 et de 9 à 12 semaines de grossesse, lorsque le placenta est formé. La maladie, l'exposition aux produits chimiques ou aux rayonnements d'une femme enceinte pendant ces périodes peut entraîner des malformations intra-utérines du fœtus.
La troisième période critique de la grossesse est de 18 à 22 semaines, lorsque la pose des connexions neuronales du cerveau se produit et que le système hématopoïétique commence son travail. Cette période est associée à un retard mental du fœtus.
Facteurs de risque d'anomalies fœtales
Facteurs de risque maternels pour CM :
- âge supérieur à 35 ans - retard de croissance intra-utérin, troubles génétiques;
- jusqu'à 16 ans - prématurité, manque de vitamines et de minéraux;
- statut social bas - infections, hypoxie fœtale, prématurité, retard de croissance intra-utérin;
- manque d'acide folique - malformations congénitales du système nerveux;
- alcool, drogues et tabagisme - retard de croissance intra-utérin, syndrome de mort subite, syndrome d'alcoolisation fœtale ;
- infections (varicelle, rubéole, infections herpétiques, toxoplasmose) - malformations congénitales, retard de croissance intra-utérin, pneumonie, encéphalopathie;
- hypertension artérielle - retard de croissance intra-utérin, asphyxie;
- polyhydramnios - malformations congénitales du système nerveux central, pathologies du tractus gastro-intestinal et des reins;
- maladies thyroïdiennes - hypothyroïdie, thyrotoxicose, goitre;
- maladie rénale - retard de croissance intra-utérin, néphropathie, mortinatalité;
- maladies des poumons et du cœur - malformations cardiaques congénitales, retard de croissance intra-utérin, prématurité;
- anémie - retard de croissance intra-utérin, mortinatalité;
- saignement - anémie, prématurité, mortinaissance
Facteurs de risque de malformations congénitales du fœtus :
- anomalies de la présentation fœtale - hémorragie, malformations congénitales, traumatismes ;
- grossesse multiple - transfusion fœto-fœtale, asphyxie, prématurité ;
- retard de croissance intra-utérin - mortinaissance, malformations congénitales, asphyxie,
Facteurs de risque lors de l'accouchement : - naissance prématurée - lourde de développement de l'asphyxie;
- accouchement tardif (accouchement retardé de 2 semaines ou plus) - le développement d'une asphyxie ou d'une mortinaissance est possible ;
- accouchement prolongé - asphyxie, mortinatalité;
- prolapsus du cordon ombilical - asphyxie.
Anomalies dans le développement du placenta :
- petit placenta - retard de croissance intra-utérin;
- gros placenta - développement de l'hydropisie du fœtus, insuffisance cardiaque;
- décollement prématuré du placenta - une perte de sang importante est possible, le développement d'une anémie;
- placenta praevia - lourd de pertes de sang et de développement d'anémie.
Diagnostic des malformations fœtales
Le diagnostic prénatal des anomalies fœtales et des pathologies génétiques est un processus très complexe. L'une des étapes de ce diagnostic consiste en des examens de dépistage prescrits à une femme enceinte pour une période de 10-12, 20-22 et 30-32 semaines (dans chacun des trimestres). Cette analyse est un test sanguin pour les marqueurs sériques biochimiques de la pathologie chromosomique (malformations).
Cela permettra d'obtenir une hypothèse sur la présence ou l'absence d'anomalies chromosomiques chez le fœtus, et une échographie en tant que méthode de diagnostic supplémentaire montrera s'il existe des écarts dans le développement physique du fœtus. L'échographie doit être effectuée par un spécialiste hautement qualifié et sur un équipement de haute qualité. Les résultats de chaque étude sont évalués conjointement, sans séparation les uns des autres.
Le dépistage ne garantit pas une pathologie à 100%, il permet uniquement d'identifier un groupe à haut risque parmi les femmes enceintes. Il s'agit d'une mesure importante et nécessaire et, malgré le caractère volontaire, la plupart des femmes enceintes le comprennent. Il n'est pas rare que les spécialistes aient du mal à répondre à la question de la présence d'anomalies génétiques chez le fœtus. Ensuite, selon le trimestre de la grossesse, la patiente se voit prescrire méthodes de recherche invasives :
- (étude des villosités choriales)
Elle se fait au 1er trimestre de la grossesse (11-12 semaines) et permet d'identifier des anomalies génétiques dans le développement du fœtus.
- amniocentèse (examen du liquide anatomique dans lequel se trouve le fœtus)
Au 1er trimestre, cette analyse révèle une hyperplasie du cortex surrénalien, au 2ème - maladies du SNC, pathologies chromosomiques.
- placentocentèse (examen des particules placentaires)
Elle est pratiquée de 12 à 22 semaines de grossesse pour détecter des pathologies génétiques.
- (prélèvement sanguin du cordon ombilical du fœtus)
Permet d'identifier la susceptibilité du fœtus aux maladies génétiques ou infectieuses.
Les femmes enceintes sont envoyées pour une consultation obligatoire avec un généticien :
- dont l'âge dépasse 35 ans;
- avoir un enfant ou des enfants atteints de troubles génétiques ;
- qui avaient des antécédents de fausses couches, de grossesses non évolutives, de mortinatalité ;
- dans la famille de qui il y a des parents atteints du syndrome de Down et d'autres anomalies chromosomiques ;
- récupéré de maladies virales au 1er trimestre de la grossesse;
- prendre des médicaments interdits pendant la grossesse;
- exposé aux radiations.
Pour le diagnostic des pathologies fœtales après la naissance, les méthodes de recherche suivantes : analyses de sang, d'urine et d'autres fluides biologiques, rayons X, imagerie par résonance magnétique et informatisée, échographie, angiographie, broncho et gastroscopie, autres méthodes immunitaires et moléculaires ...
Indications pour l'interruption de grossesse
Toute détection de malformations fœtales implique une proposition d'interruption de grossesse pour des raisons dites médicales. Si une femme refuse et décide de garder l'enfant, elle est placée sous contrôle spécial et la grossesse est surveillée plus attentivement.
Mais la future mère doit comprendre que non seulement ses sentiments et ses expériences sont importants ici, mais aussi le fait que les enfants nés avec de graves malformations et pathologies s'avèrent souvent non viables ou restent gravement handicapés à vie, ce qui, bien sûr, est très difficile pour toute famille.
Il existe d'autres indications à l'avortement:
- néoplasmes malins (la grossesse avec cancer est contre-indiquée);
- maladies du système cardiovasculaire (malformations cardiaques, thrombose veineuse profonde, thromboembolie);
- maladies neurologiques (sclérose en plaques, myasthénie grave);
- maladies infectieuses (, sous une forme active, à un stade aigu et sévère,);
- maladies du sang et des organes hématopoïétiques (hémoglobinopathie, anémie aplasique, leucémie);
- maladies oculaires (maladies du nerf optique et de la rétine);
- maladie rénale (lithiase urinaire aiguë et avec gros calculs, aiguë);
- maladies diffuses du tissu conjonctif;
- troubles endocriniens (, thyrotoxicose, hypothyroïdie non compensée dans les formes sévères) ;
- certaines maladies gynécologiques;
- indications obstétricales (insensibles au traitement et sévères, accompagnées de vomissements sévères, maladie trophoblastique gestationnelle, maladies héréditaires sévères détectées pendant la grossesse, etc.)
L'avortement pour raisons médicales n'est pratiqué qu'avec le consentement de la patiente.
Prévention des malformations congénitales du fœtus
La principale mesure visant à prévenir la survenue de malformations congénitales du fœtus est la planification de la grossesse. Non seulement le succès de la conception, mais aussi le processus de réalisation d'une grossesse, un accouchement rapide et correct et la santé de la mère et de l'enfant à l'avenir peuvent dépendre d'une préparation de haute qualité.
Avant de planifier une grossesse, il est nécessaire de subir une série d'examens : faire des tests de dépistage (MST), VIH, hépatite, syphilis, vérifier la coagulation du sang, le statut hormonal, assainir la cavité buccale, faire une échographie des organes pelviens pour exclure les inflammations maladies et néoplasmes, consultez un thérapeute pour identifier toutes les maladies chroniques possibles, idéalement les deux parents devraient être testés génétiquement.
Le point clé de la prévention des anomalies congénitales du fœtus est le maintien d'un mode de vie sain, le rejet des mauvaises habitudes, une alimentation équilibrée et nutritive et l'exclusion de l'impact sur votre corps de tout facteur négatif et nocif. Pendant la grossesse, il est important de traiter toutes les maladies possibles en temps opportun et de suivre les instructions de l'obstétricien-gynécologue.
Traitement des malformations congénitales du fœtus
Les méthodes de traitement des malformations congénitales du fœtus varient considérablement selon la nature et la gravité de l'anomalie. Les statistiques sur cette question ne sont malheureusement pas encourageantes. Un quart des enfants atteints d'anomalies congénitales meurent au cours de la première année de vie.
Un autre 25% peut vivre assez longtemps, tout en ayant des troubles physiques et mentaux intraitables ou difficiles à traiter. Et seuls 5 % des enfants nés avec des malformations congénitales peuvent être traités, le plus souvent chirurgicalement. Dans certains cas, un traitement conservateur aide. Parfois, les malformations ne deviennent perceptibles qu'en vieillissant, certaines sont complètement asymptomatiques.







